Received: октябрь 2012
Fluorine Notes, 2014, 97, 1-2
Химия и технология полифторированных органических соединений на основе новых агрессивостойких катализаторов
В.Ю. Захаров
Федеральное государственное бюджетное образовательное учреждение высшего профессионального образования«Вятский
государственный университет» (ФГБОУ ВПО «ВятГУ»), 610000, г. Киров, ул. Московская 36
e-mail:
zakhar.05@mail.ru
Аннотация. Разработаны научные принципы подбора эффективных катализаторов прямого газофазного фторирования органических соединений и экспериментально показана возможность резкого увеличения селективности и скорости фторирования неразбавленным фтором при направленном изменении природы каталитической композиции. Применение созданных катализаторов позволило усовершенствовать существующие и разработать новые ресурсосберегающие, экологически более чистые технологии целого ряда полифторированных органических продуктов.
Ключевые слова: Фторорганические соединения, фтор, гетерогенный катализатор, каталитическое фторирование, изомеризация.
окончание
3.3. Окисление тетрафторэтилена; получение карбонилдифторида
Карбонилдифторид (КФ) является основой для синтеза целого ряда килородсодержащих фтоорганических соединений, в частности, перфторметилперфторвинилового эфира /138/. Одним из перспективных путей получения КФ является окисление ТФЭ молекулярным кислородом:
C2F4 + O2 → 2COF2 – 620 кДж/моль
Газофазное взаимодействие ТФЭ и кислорода сопровождается выделением значительных количеств тепла и носит взрывной характер; это существенным образом затрудняет осуществление процесса как в лабораторном, так и в промышленном масштабах.
Одним из возможных путей для предотвращения взрывной реакции является использование инертного разбавителя. В качестве разбавителя применяют КФ /241/ – 20-100 молей на моль кислорода; процесс осуществляют при 470-720 К, времени контакта 1-10 сек и мольном отношении ТФЭ:O2, равном 1. Отмечается /241,242/, что основным продуктом такого высокотемпературного газофазного окисления является КФ; так, при 694 К конверсия ТФЭ составила 99,5%, а выход КФ – 98% /241/. В качестве инертного разбавителя при газофазном окислении ТФЭ кислородом описаны также дифторхлорметан к трифтортрихлорэтан /243/.
Отметим, что осушествление процесса с добавкой низкокипящих инертных разбавителей хотя и повышает взрывобезопасность окисления, существенным образом затрудняет выделение целевого продукта; рецикл КФ в качестве инертного разбавителя также может сопровождаться его потерями. Неприемлемым здесь является и разработанный нами взрывобезопасный способ окисления фторорганических соединений в водородо-воздушном пламени /244,245/, так как наличие воды приведет к гидролизу и потере части целевого продукта.
Выше нами была показана высокая эффективность α-Al2O3 в качестве носителя для катализаторов широкого класса термических превращений полифторированных органических соединений. В этой связи нами была изучена возможность получения КФ окислением ТФЭ неразбавленным кислородом в слое α-Al2O3, модифицированного оксидами никеля, меди, железа, кобальта. Результаты приведены в табл. 36; здесь же, для сравнения, предоставлены данные, полученные при окислении ТФЭ в слое кварцевой насадки.
Приведенные результаты свидетельствуют о существенном влиянии природы катализатора на скорость окисления. Так, на кварцевой насадке окисление ТФЭ начинается при 460-470 К, в то время как при использовании α-Al2O3, модифицированном медью, никелем и железом, а также на немодифицированном α-Al2O3 температура зажигания реакции снижается до 350-360 К. Окисление протекает при этом без индукционного периода и характеризуется практически количественным образованием КФ.
Повышенной активностью обладает α-Al2O3, модифицированный кобальтом; окисление ТФЭ в этом случае нацело протекает при начальной температуре в реакторе всего 323 К (оп. 17, табл. 36).
Интересной особенностью всех исследованных катализаторов является снижение температуры зажигания окисления после эксплуатации. Анализ состава гетерогенных контактов (после термической обработки смесью ТФЭ и O2) показал наличие на их поверхности перфторированных полипероксидов (ПС), которые, по-видимому, являются инициаторами окисления и обуславливают увеличение активности каталитических систем (табл. 37).
Таблица 36. Окисление тетрафторэтилена кислородом (объемная скорость подачи реагентов 50 час-1, мольное отношение ТФЭ:О2=1).
|
# оп |
Катализатор |
Темпе-ратура, К |
Состав реакционных газов1, объемн. % |
|||||
|
CF4 |
CO2 |
COF2 |
C2F4 |
CF3COF |
Прочие |
|||
|
1 |
SiO2 (кварц) |
453 |
0,01 |
0,02 |
0,80 |
99,12 |
0,04 |
0,02 |
|
2 |
То же |
463 |
0,01 |
0,04 |
46,38 |
52,04 |
0,30 |
1,24 |
|
3 |
-«- |
473 |
<0,01 |
0,08 |
95,31 |
2,42 |
0,84 |
1,35 |
|
4 |
α-Al2O3 |
343 |
<0,01 |
0,02 |
0,74 |
99,20 |
0,02 |
0,02 |
|
5 |
То же |
353 |
<0,01 |
0,04 |
89,32 |
9,22 |
0,62 |
0,80 |
|
6 |
-«- |
363 |
0,53 |
0,80 |
95,62 |
0,82 |
1,52 |
0,71 |
|
7 |
Ni/α-Al2O3 |
343 |
<0,01 |
0,02 |
1,94 |
97,71 |
0,02 |
0,31 |
|
8 |
То же |
353 |
<0,01 |
0,08 |
92,46 |
6,22 |
0,80 |
0,44 |
|
9 |
-«- |
363 |
<0,01 |
0,08 |
96,98 |
1,25 |
1,16 |
0,53 |
|
10 |
Cu/ α-Al2O3 |
343 |
0,01 |
0,02 |
0,68 |
99,02 |
0,02 |
0,28 |
|
11 |
То же |
353 |
0,17 |
0,36 |
72,81 |
19,24 |
6,98 |
0,44 |
|
12 |
-«- |
363 |
0,26 |
0,40 |
86,30 |
5,38 |
7,12 |
0,54 |
|
13 |
Fe/ α-Al2O3 |
343 |
0,02 |
0,04 |
0,98 |
98,62 |
0,16 |
0,18 |
|
14 |
То же |
353 |
0,12 |
0,26 |
87,87 |
9,62 |
1,60 |
0,53 |
|
15 |
-«- |
373 |
0,24 |
0,35 |
94,94 |
0,80 |
3,05 |
0,62 |
|
16 |
Со/ α-Al2O3 |
313 |
<0,01 |
0,04 |
0,09 |
99,77 |
0,02 |
0,08 |
|
17 |
То же |
323 |
<0,01 |
0,08 |
98,04 |
0,10 |
1,68 |
0,10 |
|
18 |
-«- |
333 |
<0,01 |
0,08 |
98,17 |
<0,01 |
1,62 |
0,13 |
1 Без учета непрореагировавшего кислорода.
Обработанный катализатор Со/α-Al2O3 обладает активностью уже при комнатной температуре даже после длительного хранения на воздухе (оп 6, табл. 37). Это обстоятельство, наряду с отсутствием индукционного периода, весьма важно с практической точки зрения и обеспечивает повышение взрывобезопасности процесса, так как исключает возможность неполной конверсии реагентов и соответствующее возникновение взрывоопасных смесей (в коммуникациях) даже при отключении обогрева реактора.
Таблица 37. Влияние термической (473 К) обработки смесью тетрафторэтилена и кислорода на свойства катализаторов (время обработки – 24 часа).
|
# оп. |
Катализатор |
Температура зажигания эквимолярной смеси ТФЭ и О2, К |
Содержание ПС, г-экв на 100 г катализатора, х 103 |
|
|
для свежего образца |
для обработанного образца |
|||
|
1 |
Кварц |
460-470 |
420-430 |
- |
|
2 |
α-Al2O3 |
350-360 |
320-330 |
13,0 |
|
3 |
Ni/α-Al2O3 |
350-360 |
320-330 |
1,7 |
|
4 |
Cu/α-Al2O3 |
350-360 |
320-330 |
0,3 |
|
5 |
Fe/ α-Al2O3 |
350-360 |
320-330 |
0,3 |
|
6 |
Со/ α-Al2O3 |
320-325 |
<298 |
0,4 |
Особенностью катализаторов на основе α-Al2O3 является, как показали испытания, их высокая стабильность: эксплуатация в течение 400 часов не приводит к снижению степени конверсии, уменьшению механической прочности образцов и характеризуется практически количественным выходом КФ. По результатам этой части работы выданы исходные данные на проектирование технологии КФ окислением ТФЭ кислородом на катализаторе оксид кобальта/α-Al2O3 (марки КГН-І).
Отметим, что результаты этой части работы, изложенные в /246/, побудили исследования по подбору гетерогенных катализаторов окисления ТФЭ /142/. В результате этих работ были созданы катализаторы, представляющие собой ПС на поверхности твердого носителя – активированного угля, силикагеля или алюмогеля, обладающие повышенной активностью даже в мягких условиях. Недостатком этих катализаторов является их постепенное разрушение при длительной эксплуатации, что обусловлено использованием носителей, неустойчивых в окислительных фторирующих средах.
Таким образом, на основе обобщения результатов исследования широкого класса термических газофазных превращения полифторированных органических соединений установлено, что α-Al2O3 является универсальным агрессивостойким носителем для эффективных катализаторов этих процессов. На основе α-Al2O3 создан универсальный эффективный катализатор прямого фторирования полифторированных органических соединений (NiF2/α-Al2O3), стабильные катализаторы селективного дехлорирования водородом 1,2-дихлоргексафторпропана до ГФП (Ni/α-Al2O3), 1,2-дифтортетрахлорэтана до 1,2-дифтордихлорэтилена (α-Al2O3; Ni/α-Al2O3), 1,1,2-трифтортрихлорэтана до трифторхлорэтилена (Cu, BaO/α-Al2O3), разработан низкотемпературный катализатор направленного окисления ТФЭ до КФ (оксид кобальта/α-Al2O3).
4. Катализ активированным углем нуклеофильных термических реакций полифторированных органических соединений
Активированный уголь (АУ) весьма устойчив к действию фторорганических соединений, фтористого, а также хлористого водорода и не разрушается этими агрессивными продуктами даже при длительном контактировании и повышенной температуре. Это создает предпосылки для использования активированного угля в качестве катализатора термических превращений полифторированных органических соединений.
Активированный уголь используют, как правило, в качестве носителя для приготовления электрофильных катализаторов газофазного фторирования фтористым водородом, диспропорционирования или изомеризации фторхлоруглеродов. Так, активированный уголь, модифицированный оксидами, оксифторидами, фторидами или хлоридами хрома, молибдена, железа, алюминия или никеля применяют при фторировании фтористым водородом хлороформа /247,248/, 1,1,2-трифтор-2-хлорэтилдихлорметилового эфира /249/, β-трихлорметилпиридина /250/, метилового спирта /251/; оксид хрома, нанесенный на активированный уголь, в соответствии с /252/, является эффективным катализатором диспропорционирования 1,1,1-трифтортрихлорэтана и 1,1,1-трифтор-2-хлорэтана, а активированный уголь, модифицированный солями хрома, железа, молибдена и меди – катализатором изомеризации 1,1-дихлортетрафторэтана /253/.
В /254-256/ описано использование активированного угля в качестве носителя для фторидов щелочных металлов – нуклеофильных катализаторов изомеризации оксидов тетрафторэтилена /254/ и гексафторпропилена /255,256/. Во всех этих работах описан модифицированный активированный уголь, что затрудняет оценку природы каталитического действия непосредственно углеродной поверхности из-за искажающего влияния нанесенного активного компонента.
Значительно меньше работ посвящено катализу реакций фторорганических соединений на немодифицированном угле. Так, длительное контактирование активированного угля с гексафторпропиленоксидом при 260-300 К (40-70 часов) или с тетрафторэтиленоксидом при 200-310 К /257-262/ приводит к образованию соответствующих олигомерных перфторполиэфиров с широким молекулярно-массовым распределением. В /263/ описано получение 2-перфторметилперфторпентена-2 при нагревании гексафторпропилена в присутствии активированного угля. Характерно, что все эти реакции – олигомеризация тетрафторэтиленоксида, гексафторпропиленоксида и гексафторпропилена, являются типичными нуклеофильными процессами, которые интенсифицируются основными гетерогенными контактами – галогенидами /264-267/ и перфторалкоксидами щелочных металлов /268/, солями одновалентной меди /269,270/, солями четвертичных аммониевых и фосфониевых оснований /266/. Это дает основание полагать, что и немодифицированный активированный уголь также является основным катализатором нуклеофильного типа.
Активированный уголь широко используется в химической промышленности как адсорбент и катализатор, в частности, окисления диоксида серы и сероводорода, синтеза фосгена и хлористого сульфурила, тримеризации хлорциана /271/. Учитывая доступность активированного угля, его высокую удельную поверхность и механическую прочность, представлялось интересным более подробно изучить каталитические свойства этого контакта в нуклеофильных реакциях полифторированных органических соединений, ряд которых, как показал предварительный аналитический обзор, интенсифицируется активными центрами углеродной поверхности. Особый интерес здесь представляли газофазные реакции, для которых менее характерны диффузионные ограничения, присущие катализу на узкопористых контактах, в частности, на активированном угле.
В качестве модельной реакции для оценки нуклеофильной каталитической активности угля нами была изучена газофазная изомеризация гексафторпропиленоксида (ОГФП) до перфторпропионилфторида (ПФ), которая интенсифицируется типичными основными катализаторами – фторидами щелочных металлов /255, 272/, вторичными и третичными аминами (алифатическими и ароматическими), например, триметиламином, триэтиламином, диметиламином, пиридином /273-275/, а также третичными амидами, например, диметилформамидом, диметилацетамидом, диэтилбензамидом /272/. Отметим, что изучение изомеризации ОГФП имеет и практический интерес, так как на основе ПФ может быть синтезирован соответствующий диацильный пероксид – эффективный инициатор сополимеризации фторолефинов /276/.
4.1. Изомеризация гексафторпропиленоксида до перфторпропионилфторида
Предварительные испытания тщательно дегидратированного немодифицированного активированного угля АР-В обнаружили высокую каталитическую активность в изомеризации ОГФП до ПФ: так, при 423 К и времени контакта 10 сек наблюдалась полная конверсия ОГФП.
Для количественной оценки активности нуклеофильных центров углеродной поверхности представлялось интересным сопоставить кинетические характеристики изомеризации ОГФП на активированном угле и типичных нуклеофильных катализаторах – фторидах щелочных металлов. В этой связи нами были приготовлены и испытаны дегидратированные фториды цезия, калия и натрия; эти опыты выявили низкую эффективность индивидуальных фторидов щелочных металлов, которая была обусловлена их невысокой активностью и, главное, механическим разрушением в ходе процесса, что приводило к резкому увеличению сопротивления слоя катализатора.
Известно, что диспергирование активного компонента на носителе в ряде случаев позволяет существенно увеличить эффективность каталитического действия, что обусловлено, в основном, ростом удельной поверхности активной фазы /277/; использование механически прочного носителя обеспечивает в этом случае высокую прочность нанесенного катализатора.
В этой связи нами были приготовлены и испытаны нанесенные катализаторы – фториды цезия, калия и натрия, диспергированные на поверхности плавленого фторида кальция, который обладает высокой механической прочностью и, кроме того, устойчив во фторирующих средах. Важным здесь является также и то, что этот носитель не обладает электрофильной активностью, способствующей протеканию побочной реакции – изомеризации ОГФП до перфторацетона (ПФА) /272, 278-284/.
Данные по каталитическим свойствам нанесенных фторидов щелочных металлов, а также немодифицированного, исходного плавленого фторида кальция приведены в табл. 38, здесь же представлены результаты по каталитическим свойствам предварительно дегидратированного активированного угля.
Основными продуктами превращения ОГФП на немодифицированном фториде кальция являются перфторацетилфторид и тетрафторэтилен; степень конверсии при 483 К в этом случае составила всего 10,7 %. Модифицирование фторида кальция фторидами щелочных металлов приводит к резкому увеличению скорости изомеризации; так, нанесенные фториды цезия и калия проявляют заметную активность уже при 410-430 К, а при 470-480 К наблюдается практически полное превращение ОГФП, выход ПФ при этом достигает 99,6 %.
Предварительно дегидратированный активированный уголь (АУ) обладает исключительно высокой начальной активностью в изомеризации ОГФП и позволяет осуществить полную конверсию субстрата уже при 373 К. Характерной особенностью каталитического действия активированного угля является невысокая стабильность действия при относительно низких температурах (340-470 К), что обусловлено, по-видимому, образованием олигомеров ОГФП (см. табл. 38); адсорбция высокомолекулярных олигомеров на активных центрах катализатора может привести к их блокировке и, соответственно, уменьшению активности угля, отвечающий стационарному состоянию его поверхности, устанавливается примерно через 2-4 часа после начала эксплуатации.
Таблица 38. Каталитическая изомеризация гексафторпропиленоксида (объемная скорость подачи субстрата – 250 час-1)
|
# оп. |
Катализатор1 |
Температура, К |
Степень конверсии ОГФП, % |
Выход продуктов, масс. % |
|||
|
Изомеризации |
Деструкции |
Олигомеризации |
|||||
|
ПФ |
ПФА |
||||||
|
1 |
CaF2 |
483 |
10,7 |
24,2 |
<0,1 |
75,8 |
<0,1 |
|
2 |
То же |
573 |
98,2 |
2,6 |
<0,1 |
97,4 |
<0,1 |
|
3 |
NaF/CaF2 |
433 |
8,4 |
99,9 |
<0,1 |
0,1 |
<0,1 |
|
4 |
То же |
473 |
84,4 |
99,4 |
<0,1 |
0,6 |
<0,1 |
|
5 |
KF/CaF2 |
413 |
12,5 |
99,9 |
<0,1 |
0,1 |
<0,1 |
|
6 |
То же |
478 |
99,2 |
99,5 |
<0,1 |
0,5 |
<0,1 |
|
7 |
CsF/CaF2 |
413 |
18,7 |
99,9 |
<0,1 |
0,1 |
<0,1 |
|
8 |
То же |
473 |
99,2 |
99,6 |
<0,1 |
0,4 |
<0,1 |
|
9 |
АУ2 |
373 |
97,8 |
93,7 |
2,0 |
0,3 |
4,0 |
|
10 |
То же |
473 |
99,5 |
95,0 |
3,4 |
0,8 |
0,8 |
1 Количество нанесенного фторидов натрия, калия и цезия составляло 6,6 ммоль (для каждого) на 100 г катализатора.
2 Уголь для уменьшения электофильной активности его зольной части предварительно обрабатывали водным раствором гидроксида натрия (20 масс.%) при 373 К в течение 5 часов и промывали водой; приведенные данные отвечают начальному периоду активности угля (через 1 час после начала опыта).
Характерно, что модифицирование активированного угля фторидами цезия и калия не приводит, как показали испытания, к увеличению каталитической активности; стабильность действия при этом даже несколько уменьшается.
Изучение кинетики изомеризации ОГФП на свежем активированном угле (по начальным скоростям реакции), угле после стабилизации его активности во времени, а также фторидах щелочных металлов, нанесенных на фторид кальция, показало, что в области начальных концентраций ОГФП 6,7х10-4 – 2,1х10-3 моль/л, температур 383-573 К и степеней конверсии до 12 % эта реакция протекает по первому порядку.
Кинетическая схема изомеризации ОГФП может быть представлена в следующем виде:
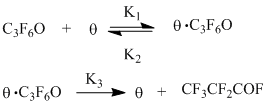
Эта схема включает обратимую адсорбцию ОГФП на свободных каталитических центрах Θ (уравнение 1) и последующую изомеризацию с образованием ПФ и регенерацией активных центров (уравнение 2).
Скорость реакции (W) в соответствии с приведенной схемой будет описываться уравнениями:
![]() (квазиравновесное
приближение)
(квазиравновесное
приближение)
![]() (квазистационарное
приближение)
(квазистационарное
приближение)
где в = К1/К2 – адсорбционный коэффициент (константа равновесия адсорбции) ОГФП, С – концентрация ОГФП.
При малых значениях концентрации ОГФП (вС<<1) в квазиравновесном приближении W=К3вС=Кэфф.С, что согласуется с экспериментально определенным первым порядком скорости реакции по концентрации ОГФП.
Данные по температурным зависимостям констант скоростей изомеризации ОГФП для изученных катализаторов приведены на рис. 9; в табл. 39 приведены значения соответствующих предэкспоненциальных множителей и энергий активаций.
Энергия активации изомеризации ОГФП на свежем активированном угле в интервале температур 443-543 К составляет всего 15,1 кДж/моль, что свидетельствует о протекании реакции во внутреннедиффузионной области /285/. При температурах 383-413 К реакция переходит, по-видимому, в кинетическую область, о чем свидетельствует резкое увеличение энергии активации, которая составляет в этом случае 50,2 кДж/моль. Интересно, что энергия активации изомеризации на отработанном активированном угле (после стабилизации его активности во времени) составляет во всем исследованном интервале температур 56,5 кДж/моль и близка к значению энергии активации реакции на свежем активированном угле в кинетической области.
Энергия активации изомеризации ОГФП закономерно уменьшается в ряду нанесенных фторидов натрия, калия и цезия (93,3; 81,6 и 55,6 кДж/моль, соответственно), что свидетельствует об увеличении активности фтор-иона с ростом радиуса катиона щелочного металла и согласуется с результатами многочисленных исследований по изучению нуклеофильной активности иона фтора /286,287/.
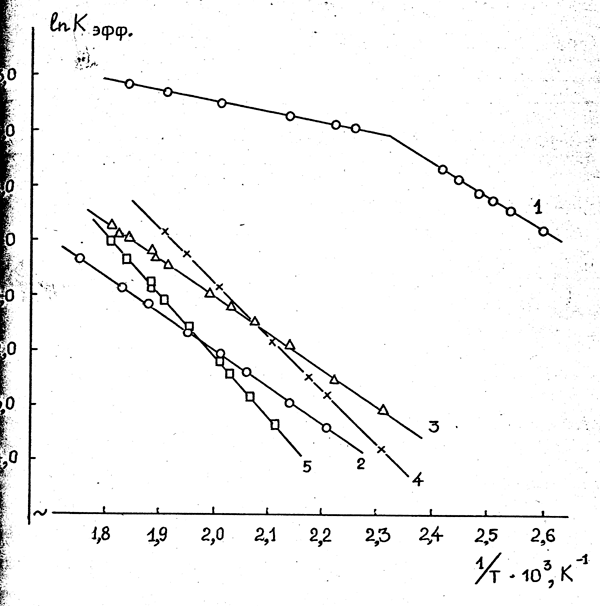
Рисунок 9. Температурные зависимости (в аррениусовых координатах) констант скоростей (Кэфф.) изомеризации гексафторпропиленоксида до перфторпропионилфторида на свежем (1) и отработанном (2) активированном угле, а также на фторидах цезия (3), калия (4) и натрия (5), нанесенных на фторид кальция.
Таблица 39. Кинетические характеристики каталитической изомеризации гексафторпропиленоксида до перфторпропионилфторида.
|
# оп. |
Катализатор |
Температурный интервал, К |
lg Ко |
Еакт., кДж/моль |
|
1 |
Активированный уголь (свежий) |
383-413 |
6,9±0,4 |
50,2±3,3 |
|
2 |
То же |
443-543 |
2,6±0,1 |
15,1±0,8 |
|
3 |
Активированный уголь (отработанный) |
453-573 |
5,0±0,1 |
56,5±0,8 |
|
4 |
CsF/CaF2 |
433-553 |
5,3±0,1 |
55,6±0,8 |
|
5 |
KF/CaF2 |
433-523 |
8,2±0,1 |
81,6±0,8 |
|
6 |
NaF/CaF2 |
473-553 |
8,8±0,1 |
93,3±0,8 |
Интересно, что энергия активации изомеризации ОГФП на активированном угле и нанесенном фториде цезия весьма близки (50,2 и 55,6 кДж/моль, соответственно), что свидетельствует о высокой нуклеофильных активности каталитических центров углеродной поверхности. Этот результат побудил дальнейшие работы по изучению каталитических свойств активированного угля в практически важных реакциях фторорганических соединений, протекающих по нуклеофильному типу.
Отметим, что по результатам этой части работы выданы исходные данные на проектирование опытно-промышленной установки по получению перфторпропионилфторида каталитической изомеризацией гексафторпропиленоксида.
4.2. Селективная термодеструкция олигомеров карбонилдифторида и перфторированных полипероксидов в присутствии перфторполиоксаметиленацетилфторидов.
Низкомолекулярные перфторполиоксаметиленацетилфториды СF3O(CF2O)nCF2COF со средним молекулярным весом ~ 300 (КЧФn), являющиеся ценным сырьем для получения нового поколения термоморозоагрессивостойких фторкаучуков с уникальными свойствами /287,288/, образуются при термическом жидкофазном окислении ГФП кислородом в среде 1,1,2-трифтортрихлорэтана /289/; выход КЧФn при этом, впрочем, невелик и составляет 1,8-2,0 масс. %. Нами разработана и внедрена технология получения КЧФn и ОГФП термическим жидкофазным окислением ГФП кислородом в среде 1,1,2-трифтортрихлорэтана с добавками селективного инициатора – 1,2-дибромтетрафторэтана /290/, позволившая существенно повысить скорость процесса /291/, увеличить (в 1,5 раза) выход КЧФn /292/ и ОГФП – с 36-37 до 48-49% /293/, а также снизить удельный вклад деструктивного окисления («горения») ГФП /294/, которое протекает с образованием побочных продуктов – карбонилдифторида и ацетилфторида.
Наряду с КЧФn в процессе окисления ГФП образуются олигомеры карбонилдифторида с концевой фторформиатной группой СF3O(CF2O)nCOF (ОКФ) и перфторированные полипероксиды (ПС), которые накапливаются в растворителе и затрудняют транспортировку и выделение КЧФn. Это обуславливает необходимость предварительной очистки растворов КЧФn от ОКФ и ПС.
Имеется сообщение /295/ о возможности разрушения ОКФ и ПС при их термообработке в вакууме (470-500 К); в то же время необходимость осуществления процесса при пониженном давлении создаст дополнительные трудности при промышленной реализации такого способа очистки.
В /296/ разработан двухстадийный способ очистки, который включает контактирование раствора (на первой стадии) в периодическом режиме с катализатором разложения ОКФ – безводным фтористым калием и затем, на второй стадии – термическую обработку инициатором разложения ПС – диметиловым эфиром диэтиленгликоля (диглимом). Такой двухстадийный способ хотя и позволяет достичь высокой степени очистки КЧФn от ОКФ и ПС, весьма сложен, характеризуется повышенной энерго- и материалоемкостью, низкой производительностью и наличием отходов – отработанных фтористого калия и диглима. В то же время, приведенные в /296/ данные указывают на возможность интенсификации разложения ОКФ катализаторами нуклеофильного типа. В этой связи нами была изучена газофазная термодеструкция ОКФ и ПС в смеси с КЧФn на предварительно дегидратированном активированном угле, который, как отмечалось выше, является перспективным катализатором нуклеофильного типа. Осуществление термодеструкции в газовой фазе на неподвижном слое твердого катализатора имеет очевидные технологические преимущества, в частности, позволяет организовать непрерывный процесс; при этом отпадает необходимость в дополнительной стадии отделения катализатора от продуктов реакции. Отметим, что одним из основных требований к катализатору, кроме высокой активности и стабильности в разложении как ОКФ, так и ПС, является отсутствие на нем термодеструкции целевых продуктов – КЧФn.
Данные по термодеструкции ОКФ и ПС в смеси с КЧФn на предварительно дегидратированном активированном угле приведены в табл. 40; здесь же, для сравнения представлены результаты, полученные в полом реакторе, без катализатора и на типичных катализаторах нуклеофильного типа – тщательно дегидратированных фторидах калия и натрия.
Таблица 40. Термокаталитическое разложение перфторированных полипероксидов, олигомеров карбонилдифторида и перфторполиоксаметиленацетилфторидов.1
|
# оп. |
Катализатор |
Температура, К |
Объемная скорость подачи реагентов, час-1 |
Степень разложения, % |
||
|
ПС |
ОКФ |
КЧФn |
||||
|
1 |
Без катализатора |
473 |
3600 |
11,1 |
6,2 |
0,3 |
|
2 |
То же |
873 |
3600 |
99,0 |
99,0 |
9,9 |
|
3 |
-«- |
473 |
360 |
29,7 |
19,5 |
2,9 |
|
4 |
-«- |
723 |
360 |
99,2 |
99,0 |
12,9 |
|
5 |
NaF |
473 |
3600 |
14,7 |
27,9 |
15,1 |
|
6 |
KF |
473 |
3600 |
2,3 |
61,1 |
2,9 |
|
7 |
Активированный уголь |
373 |
3600 |
85,3 |
96,4 |
0,8 |
|
8 |
То же |
373 |
360 |
99,5 |
99,0 |
1,8 |
|
9 |
-«- |
358 |
400 |
99,5 |
99,0 |
<0,1 |
|
10 |
-«- |
353 |
360 |
99,5 |
99,0 |
<0,1 |
1 Результаты оп. 9 и 10 получены в опытно-промышленных условиях
Из приведенных данных видно, что ПС, ОКФ и в особенности КЧФn являются весьма термостойкими соединениями. При температуре 573 К и объемной скорости подачи 3600 час-1 степень их разложения (в полой трубе, без катализатора) составила всего 18,4; 10,4 и 2,7 %, а при 773 К – 98,2; 46,8 и 5,7 %, соответственно.
Практически полное некаталитическое разрушении ОКФ и ПС (объемная скорость подачи реагентов – 3600 час-1) достигается лишь при 873 К. Обращает внимание исключительно высокая термическая устойчивость КЧФn, степень разложения которых в этих же условиях составляет всего 9,9 %.
Уменьшение объемной скорости подачи реагентов до 360 час-1 позволяет снизить температуру полного каталитического разложения ОКФ и ПС до 723 К; степень деструкции КЧФn составляет при этом 12,9 %.
Проведенное исследование позволило количественно оценить сравнительную термостабильность индивидуальных перфторполиоксаметиленацетилфторидов с различным числом атомов углерода (рис. 10).
Видно, что термическая устойчивость КЧФn существенно зависит от длины их цепи. Так, при 873 К и времени контакта 1 сек (объемная скорость подачи реагентов 3600 час-1) перфторполиоксаметиленацетилфториды с n = 1 практически не разрушаются; степень же разложения КЧФn с n = 2, 4, 6 и 9, например, составляет 6,0; 9,0; 12,6 и 19,0 %, соответственно.
Данные по относительной термической устойчивости ПС, ОКФ и КЧФn (табл. 40, оп. 1-4) указывают на принципиальную возможность термической некаталитической очистки перфторполиоксаметиленацетилфторидов. Недостатком такого способа является необходимость осуществления процесса при высоких температурах (720-870 К), что приведет к увеличенным энергозатратам на нагрев и охлаждение реакционной смеси. Кроме того, в условиях, обеспечивающих полное разложение ПС и ОКФ, наблюдается заметная деструкция целевых продуктов (примерно 10 %). Это обуславливает целесообразность подбора селективного бифункционального катализатора, позволяющего снизить температуру полного разложения ПС и ОКФ.
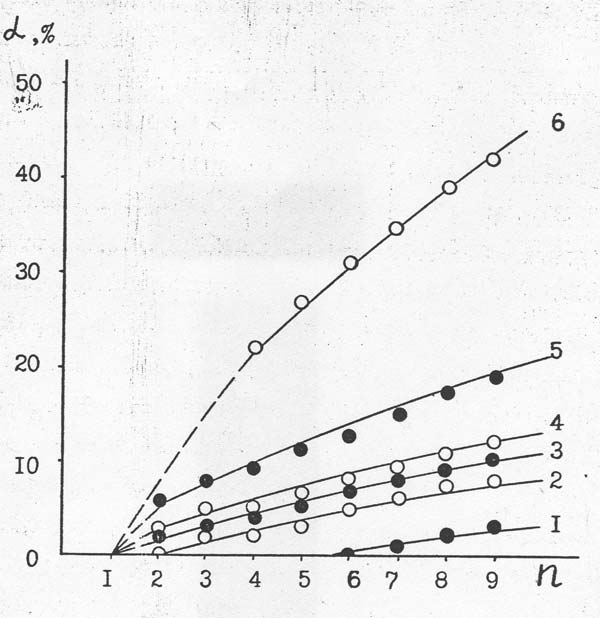
Рисунок 10. Зависимость степени термодеструкции (α) индивидуальных перфторполиоксаметиленацетилфторидов от длины их цепи (n) при объемной скорости подачи 3600 час-1 и температурах: 1 – 473 К; 2 – 573 К; 3 – 723 К; 4 – 773 К; 5 – 873 К и 6 – 923 К.
Испытание фторидов калия и натрия выявило увеличение скорости газофазного разложения ОКФ и ПС; в то же время эти катализаторы имеют недостаточно высокую стабильность. Так, начальная степень разложения ПС на фториде калия (объемная скорость подачи – 3600 час-1) при 373 К равнялась 13,8 %, тогда как через шесть часов составила всего 2,3 % даже при 473 К. Фторид калия, кроме того, механически разрушается в ходе процесса, что приводит к измельчению гранул образца и забивкам в реакторе. Разрушение катализатора может быть обусловлено его обратимым взаимодействием с фторангидридами с образованием алкоксидов, что приводит к изменению фазового состава. В случае ОКФ такие алкоксиды разлагаются с выделением карбонилдифторида и образованием мелкодисперсного фторида калия, который и способствует забивкам в реакторе.
Образцы, полученные нанесением фторида калия на CaF2 и α-AI2O3 обладают значительной механической прочностью и не разрушаются в ходе эксплуатации; в то же время активность этих контактов не высока (ниже, чем у индивидуального фторида калия) и, кроме того, уменьшается во времени.
Низкая стабильность действия может быть обусловлена блокировкой активных центров высокомолекулярными фторангидридами.
Предварительно дегидратированный активированный уголь, как следует из данных, приведенных в табл. 40, обладает исключительно высокой каталитической активностью. При 323 К и объемной скорости подачи реагентов 360 час-1 (время контакта на холодный газ – 10 сек) наблюдается практически полное разложение ПС и ОКФ (98,4 и 94,8 %, соответственно); снижение времени контакта приводит к уменьшению степени очистки. Так, при объемной скорости подачи 720 час-1 (323 К) степень очистки от ОКФ и ПС составляет 84,2 и 92,8 %, а при 1440 час-1 – 48,0 и 80,8 %, соответственно. Снижение температуры до 290-300 К при времени контакта 10 сек также приводит к уменьшению степени очистки. Из представленных данных следует, что при промышленном использовании способа для достижения высокой степени очистки температура и время контакта должны превышать 320 К и 10 сек, соответственно. Характерно, что контактирование перфторполиоксаметиленацетилфторидов с активированным углем даже в относительно жестких условиях (температура 523 К, время контакта 10 сек) не приводило к их разрушению.
Таким образом, использование катализатора – активированного угля позволяет снизить температуру полной очистки КЧФn от ПС и ОКФ с 720-870 К (для некаталитического процесса) до 320-370 К и избежать нежелательной деструкции целевых продуктов.
В заключение этой части работы отметим, что промышленные испытания подтвердили результаты лабораторных исследований (табл. 40, оп. 9, 10) и подчеркнули высокую стабильность действия активированного угля: переработка более 3 т смеси на 1 кг катализатора (эксплуатация в течение 2 лет) не привели к уменьшению его активности – степень очистки от ОКФ и ПС при этом превышала 99 %.
Промышленное внедрение способа позволило организовать систематическую переработку КЧФn, в ходе которой проведено получение новых типов термоморозоагрессивостойких фторкаучуков /287/ - СКФ-260 МПАН (в соответствии с программой «Буран») и «Фторкам» (в соответствии с программой «Наполнитель-І»).
4.3. Реакции перфторизобутилена с фтористым водородом, 1,1,5-тригидроперфторпентанолом-1 и водой
Технология синтеза ГФП – сырья для получения фторполимеров, фторкаучуков и целого ряда других фторорганических продуктов, основана на пиролизе ТФЭ. Наряду с ГФП, октафторциклобутаном (ОФЦБ), а также рядом других фторорганических продуктов (префторбутены, перфторметилперфторциклобутан и др.) при пиролизе ТФЭ образуется высокотоксичный перфторизобутилен (ПФИБ)1; поэтому для обеспечения безопасности разделения продуктов пиролиза2 их необходимо предварительно подвергать очистке от ПФИБ.
1 Предельно допустимая концентрация ПФИБ в воздухе рабочей зоны равняется 0,1 мг/м3.
2 При ректификационном разделении продуктов пиролиза кроме целевого продукта – ГФП выделяют также непрореагировавший ТФЭ и ОФЦБ (их возвращают на стадию пиролиза), и кубовые продукты, которые обезвреживают.
Характерной особенностью ПФИБ является его повышенная (по сравнению с ТФЭ и ГФП) активность в реакциях с нуклеофильными реагентами /297-303/; именно на этом свойстве основаны практически все известные способы его селективного извлечения из продуктов пиролиза ТФЭ. Так, первоначально, очистку продуктов пиролиза осуществляли селективным жидкофазным гидролизом ПФИБ до гексафторизомасляной кислоты водой в смеси с ацетоном /304/; этот способ характеризовался, впрочем, недостаточной полнотой извлечения ПФИБ, а также пожаро- и взрывоопасностью, обусловленной использованием ацетона. В настоящее время ПФИБ гидролизуют водным раствором гидроксида натрия в присутствии катализатора нуклеофильного типа – триэтиламина /305/. Этот способ обеспечивает практически полное селективное удаление ПФИБ и, кроме того, позволил организовать выделение и рецикл на пиролиз дополнительных количеств ОФЦБ и перфторбутенов (из кубовых продуктов), что привело к уменьшению удельного расхода ТФЭ, а также снижению загрязнения окружающей среды кубовыми продуктами /306,307/. Недостатком этого способа является полная минерализация ПФИБ – ценного фторорганического сырья. Учитывая значительные количества образующегося ПФИБ (130-140 кг на 1 т ГФП), актуальным является поиск путей, позволяющих наряду с полным извлечением из продуктов пиролиза осуществить также и его утилизацию.
ВАХЗ совместно с нами разработан способ утилизации ПФИБ, который заключается в контактировании продуктов пиролиза ТФЭ с 1,1,5-тригидроперфторпептанолом-1 при катализе фторидом калия /185,186/; в результате селективного взаимодействия ПФИБ с этим фторспиртом происходит образование 2,5,5,9-тетрагидро-2-перфторметил-4-оксаперфторнонана /186/. Селективное прямое фторирование этого продукта позволяет получить эффективную диэлектрическую жидкость – 2-перфторметил-4-оксаперфторнонан.
Другой путь утилизации ПФИБ может быть основан на его селективном гидрофторировании фтористым водородом с образованием моногидроперфторизобутана – эффективного рабочего тела для плазмохимической обработки элементов электронных схем /297/.
Перспективным также является изучение возможности газофазного каталитического гидролиза ПФИБ водой; применение такого способа позволило бы существенно упростить технологию дополнительной очистки кубовых продуктов.
Характерно, что все эти реакции – взаимодействие ПФИБ с фтористым водородом, 1,1,5-тригидроперфторпентанолом-1 и водой, являются процессами, протекающими по нуклеофильному типу; в этой связи нами и был изучен их катализ активированным углем.
Взаимодействие перфторизобутилена с фтористым водородом.
Данные по взаимодействию ПФИБ в составе продуктов пиролиза ТФЭ с фтористым водородом на предварительно дегидратированном активированном угле (АУ) представлены в табл. 41; здесь же, для сравнния приведены результаты, полученные с использованием полого реактора (без катализатора), а также в присутствии типичных катализаторов нуклеофильного типа – тщательно осушенных фторидов цезия и калия, нанесенных на поверхность плавленого фторида кальция.
Из приведенных данных видно, что ПФИБ в газовой фазе без катализатора не взаимодействует с фтористым водородом (при температурах до 570 К и объемной скорости подачи реагентов 80 час-1).
Контактирование ПФИБ с фтористым водородом в слое нанесенных фторидов цезия и калия (оп. 3,4, табл. 41) хотя и сопровождается образованием некоторых количеств моногидроперфторизобутана, характеризуется низкой степенью конверсии субстрата (не превышает 15%).
Использование активированного угля приводит к резкому увеличению скорости взаимодействия ПФИБ и HF (оп. 5-8, табл. 41). Так, уже при 323 К и объемной скорости подачи реагентов 750 час-1 степень конверсии ПФИБ составляет 68%; увеличение температуры до 373-423 К сопровождается полной конверсией ПФИБ (степень очистки выше 99%). Характерно, что в присутствии активированного угля фтористый водород реагирует весьма селективно – содержание в реакционных газах моногидроперфторпропана – продукта гидрофторирования ГФП, даже при 423 К не превышает 0,1 объемн.%.
Следует отметить высокую стабильность каталитического действия активированного угля – его эксплуатация в течение 400 часов (423 К, объемная скорость подачи реагентов – 80 час-1, мольное отношение HF:ПФИБ – 1,5) не приводит к уменьшению степени конверсии ПФИБ, которая в этих условиях превышает 99%.
Взаимодействие перфторизобутилена с 1,1,5-тригидроперфторпентанолом-1.
Контактирование продуктов пиролиза ТФЭ с 1,1,5-тригидроперфторпентанолом-1 на активированном угле при 473 К (оп. 10, табл. 41) также приводит к практически полной селективной конверсии ПФИБ; катализатор при этом характеризуется высокой стабильностью действия. Снижение температуры до 373 К и ниже сопровождается уменьшением полноты связывания ПФИБ фторспиртом (до 85%); активность катализатора в этом случает заметно падает во времени, что может быть обусловлено необратимой при этих температурах адсорбцией 1,1,5-тригидроперфторпентанола-1 или его высокомолекулярных аддуктов с ПФИБ на каталитических центрах углеродной поверхности.
Взаимодействие перфторизобутилена с водой.
Из приведенных в табл. 41 данных по взаимодействию ПФИБ с водой (оп. 11-14) следует, что без катализатора эта реакция практически не протекает (до 473 К). Применение нанесенного фторида калия позволяет заметно увеличить скорость гидролиза ПФИБ – при 423 К степень его конверсии на этом катализаторе составила 42%. Каталитическая активность угля, впрочем, существенно выше; степень конверсии ПФИБ в этих же условиях превышает 99%.
Характерной особенностью каталитического газофазного взаимодействия ПФИБ с водой является образование значительных количеств 2-гидроперфторпропилена (см. табл. 41), а также 2,2-дигидроперфторпропана (его содержание в реакционных газах в оп. 13,14 составило, например 0,84 и 0,43 объемн. % или 25 и 13 мольн.%, соответственно, от вступившего в реакцию ПФИБ).
Химизм образования этих примесей можно описать следующей схемой:
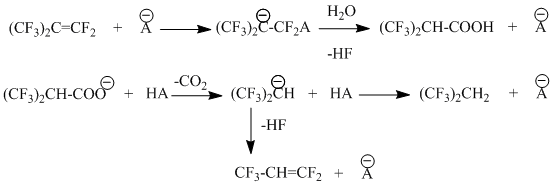
Эта схема включает в себя стадию нуклеофильной атаки ПФИБ при взаимодействии с каталитическим центром
![]() , гидролиз активированного
таким образом ПФИБ с образованием гексафторизомасляной кислоты или ее аниона, декарбокислирования
этого аниона с образованием карбаниона
, гидролиз активированного
таким образом ПФИБ с образованием гексафторизомасляной кислоты или ее аниона, декарбокислирования
этого аниона с образованием карбаниона ![]() , который либо присоединяет протон с образованием 2,2-дигидроперфторпропана,
либо, элиминируя фтор-ион, превращается в 2-гидроперфторпропилен.
, который либо присоединяет протон с образованием 2,2-дигидроперфторпропана,
либо, элиминируя фтор-ион, превращается в 2-гидроперфторпропилен.
Образование карбаниона гексафторизомасляной кислоты весьма вероятно, если учесть наличие на поверхности угля нуклеофильных центров, способствующих ее диссоциации; кроме того, появление продуктов, декарбоксиливания – C3F6H2 и C3F5H наблюдается при относительно низких температурах (300-320 К), то есть в условиях, нехарактерных для декарбокслирования гексафторизомасляной кислоты, тогда как ее анион обладает существенно меньшей устойчивостью.
Отметим, что образующийся при гидролизе ПФИБ пентафторпропилен, ухудшает качество целевого продукта – ГФП. Учитывая трудность ректификационного отделения пентафторпропилена от ГФП, целесообразным представляется предварительное разделение продуктов продуктов пиролиза ТФЭ на две фракции: лёгкую, содержащую непрореагировавший ТФЭ, ГФП и часть ОФЦБ и тяжелую, содержащую часть ОФЦБ, ПФИБ и другие высококипящие продукты пиролиза. Очистка от ПФИБ при этом следует подвергать только тяжелую фракцию, которая не содержит ГФП, что исключает возможность его загрязнения образующимся при гидиролизе ПФИБ пентафторпропиленом.
С другой стороны, одной из стадий производства ГФП, как уже отмечалось, является дополнительная очистка кубовых продуктов от непрореагировавшего ПФИБ. Эти продукты после очистки не утилизируются; осуществление этого процесса путем термического гидролиза на активированном угле позволило бы существенно упростить технологию дополнительной очистки.
Испытания показали, что контактирование кубовых продуктов с водой (количество воды составляло 10-15% от их массы) на активированном угле при 420-470 К и объемной скорости подачи реагентов 180-300 час-1 характеризуется полной очисткой от ПФИБ.
По результатам этой части работы выданы исходные данные на проектирование безотходной технологии
очистки от ПФИБ продуктов пиролиза ТФЭ (гидрофторированием) и кубовых продуктов в производстве
ГФП (гидролизом).
4.4. Термодеструкция ω–хлорперфторалкилфторсульфатов
ВАХЗ совместно с нами разработана технология утилизации α-гидро-ω–хлорперфторалканов H(CF2)nCl – отходов производства ТФЭ. /308/ В качестве промежуточных продуктов по этой технологии образуются ω-хлорперфторалкилфторсульфаты /187,188,309-311/, которые перерабатываются в соответствующие фторангидриды ω-хлорперфторкарбоновых кислот /189/ с последующим получением на их основе эффективных медицинских препаратов, пламягасящих средств, гербицидов и фторэмульгаторов /312-315/. В этой связи актуальным является подбор эффективных катализаторов селективной термодеструкции ω-хлоралкилфторсульфатов до соответствующих фторангидридов:
Cl(CF2)nOSO2F → Cl(CF2)n-1COF+SO2F2
В соответствии с /189,316/ разложению алкилфторсульфатов способствуют типичные катализаторы нуклеофильного типа – фториды щелочных металлов. В этой связи нами и была изучена возможность катализа термодеструкции ω-хлорперфторалкилфторсульфатов, в частности ω-хлорперфтороктилфторсульфата, предварительно дегидратированным активированным углем; данные представлены в табл. 42. Здесь же, для сравнения, приведены результаты по разложению ω-хлорперфтороктилфторсульфата в слое инертной насадки – плавленого фторида кальция.
Таблица 42. Каталитическая термодеструкция ω–хлорперфтороктилфторсульфата (объемная скорость подачи субстрата – 360 час-1)
|
Катализатор |
Степень конверсии Cl(CF2)8OSO2F,%, при температуре, К |
|||
|
473 |
523 |
573 |
673 |
|
|
СaF2 |
0,5 |
0,5 |
1,7 |
9,1 |
|
Активированный уголь АР-В |
6,2 |
21,5 |
52,1 |
98,2 |
Из приведенных данных видно, что активированный уголь резко увеличивает скорость термодеструкции ω-хлорперфтороктилфторсульфата; практически единственным органическим продуктом этого процесса является ω-хлорперфтороктаноилфторид.
4.5. Гидролиз 1,1,1-трифтортрихлорэтана в смеси с 1,1,2-трифтортрихлорэтаном.
1,1,1-Трифтортрихлорэтан является нежелательной примесью в 1,1,2-трифтортрихлорэтана; его наличие, в частности, уменьшает стабильность смеси изомеров хладона-113, а также сопровождается загрязнением трифторхлорэтилена, получаемого на основе 1,1,2-трифтортрихлорэтана, трудноотделяемой примесью – 1,1-дифтор-хлорэтиленом, ухудшающей качество этого фторхлорлефина. В этой связи актуальным является поиск путей очистки 1,1,2-трифтортрихлорэтана от 1,1,1-трифтортрихлорэтана, так как эти изомеры хладона-113 имеют одинаковую температуру кипения и не разделяются ректификацией.
В настоящей части работы приведены результаты по селективному нуклеофильному гидролизу малых количеств 1,1,1-трифтортрихлорэтана (0,8 масс.%) в смеси с 1,1,2-трифтортрихлорэтаном на активированном угле (табл. 43); здесь же, для сравнения, представлены данные по гидролизу этих изомеров в присутствии фторида калия (10масс.%), нанесенного на γ-Al2O3.
Таблица 43. Каталитический гидролиз 1,1,1-трифтортрихлорэтана в смеси с 1,1,2-трифтортрихлорэтаном (температура 473 К, мольное отношение вода : сумма изомеров трифтортрихлорэтана = 2).
|
# оп. |
Катализатор |
Объемная скорость подачи реагентов, час-1 |
Степень конверсии, % |
|
|
CF3-CCl3 |
CF2Cl-CFCl2 |
|||
|
1 |
γ-Al2O3 |
360 |
<0,1 |
<0,1 |
|
2 |
KF/ γ-Al2O3 |
360 |
6,2 |
1,4 |
|
3 |
Активированный уголь АР-В |
360 |
34,1 |
1,4 |
|
4 |
То же |
180 |
53,0 |
2,8 |
|
5 |
-//- |
150 |
62,0 |
3,5 |
|
6 |
-//- |
120 |
70,1 |
4,3 |
|
7 |
-//- |
90 |
82,7 |
5,6 |
Из приведенных данных видно, что скорость и селективность гидролиза на активированном угле существенно выше, чем в присутствии нанесенного фторида калия; использование активированного угля позволяет в относительно мягких условиях (температура 473 К, объемная скорость подачи реагентов – 90 час-1) осуществлять глубокий селективный гидролиз даже малых количеств 1,1,1-трифтортрихлорэтана.
4.6. Природа каталитического действия активированного угля
Выявление природы каталитического действия активированных углей является весьма сложной задачей, что обусловлено исключительно богатой химией их поверхности: кроме неорганических примесей (зола активированных углей содержит /317/ алюминий, кремний, железо, магний, кальций и калий) в карбонизированных продуктах обычно находят водород, кислород, серу и азот /318/. Все это затрудняет отнесение тех или иных каталитических свойств к какой-либо определенной группе центров.
Известно, что поверхность активированных углей содержит значительные количества хемосорбированного кислорода. Детальные исследования /318/ позволили отнести практически весь кислород, определяемый элементарным анализом, к хорошо известным функциональным группам: карбоксильной, гидроксильной (фенольной), карбонильной и лактонной. Нуклеофильные участки такой поверхности могут быть ответственными за каталитическое действие активированного угля в изученных реакциях.
В этой связи нами были проведены опыты с использованием смеси бензойной кислоты, фенола и янтарного ангидрида; функциональные группы этих соединений моделируют кислородсодержащие фрагменты угольной поверхности. Контакт такой смеси с ПС и ОКФ при 298 К, как было установлено экспериментально, не приводит к их деструкции. Здесь следует отметить, что гидроксильные, карбонильные, карбоксильные и лактонные группы поверхности угля могут обладать несколько иными свойствами, чем те же группы в составе использованной модельной смеси. Это не позволяет сделать однозначного заключения об участии кислородсодержащих фрагментов на поверхности активированного угля в нуклеофильных реакциях фторорганических соединений.
Поверхность активированного угля может содержать также свободно-радикальные центры, образующиеся на стадии активирования, которое сопровождается выгоранием части углерода кристаллитов и разрушением гексагональных колец с появлением боковых цепочек углеродных атомов. Такие радикалы, в соответствии с /318/, обладают нуклеофильными свойствами и могут ускорять изученные реакции.
Хемосорбция воды на радикальных углеродных фрагментах приводит к образованию ионо-обменных гидроксильных групп основного характера /318/. Их замещение на фтор при контакте с реакционно-способными фторорганическими соединениями может привести к возникновению каталитической системы с обладающим нуклеофильными свойствами подвижным фтор-ионом; активированный уголь при этом играет роль макро-катиона и, кроме того, способствует увеличению концентрации субстрата (за счет его повышенной адсорбционной способности) вблизи активных центров.
В заключение отметим, что исследование широкого класса термических газофазных реакций полифторированных органических соединений показало, что предварительно дегидратированный активированный уголь является эффективным универсальным катализатором нуклеофильного типа и может быть использован даже в немодифицированном виде в качестве катализатора изомеризации гексафторпропиленоксида, термодеструкции олигомеров карбонилдифторида и перфторированных полипероксидов, гидрофторирования и гидролиза префторизобутилена, а также его взаимодействия с 1,1,5-тригидроперфторпентанолом-1, разложения ω-хлорперфторалкилфторсульфатов и селективного гидролиза 1,1,1-трифтортрихлорэтана в смеси с 1,1,2-трифтортрихлорэтаном.
Доступность и простота использования этого катализатора несомненно приведут к расширению областей его практического применения.
5. ВЫВОДЫ
1. На основе теоретического обобщения результатов исследования газофазного взаимодействия широкого класса непредельных и водородсодержащих фторорганических субстратов с элементарным фтором создано новое перспективное научное направление – селективное прямое каталитическое фторирование полифторированных органических соединений.
- Разработаны научные принципы подбора эффективных агрессивостойких катализаторов прямого газофазного фторирования органических соединений и впервые экспериментально показана возможность резкого увеличения селективности и скорости фторирования неразбавленным фтором при направленном изменении природы каталитической композиции.
- Разработана эффективная универсальная каталитическая композиция – NiF2 /α-AI2O3 и с ее использованием, на примере 3-перфторизоприл-2-перфторметилперфторпентена-2 и 3-перфторизопропил-4-перфторметилперфторпентена-2 – изомеров перфторнонена, изомеров 2-перфторметилперфторпентена-3, изомеров перфторбутена, 2,5,5,9-тетрагидро-2-перфторметил-4-оксаперфторнонана, α-гидро-ω-хлорперфторалканов H(CF2)nCl (n=2÷8), а также полифторированных примесей в составе фторуглеродных смазок, перфтордекалина, перфтордиметилперфторциклогексана и других фторуглеродных жидкостей, осуществлено селективное газофазное фторирование полифторированных органических соединений с количественным выходом соответствующих практически важных продуктов селективного фторирования.
2. На основе систематического исследования широкого класса термических газофазных превращений полифторированных органических соединений установлено, что α-AI2O3 является универсальным, агрессивостойким носителем для эффективных катализаторов термического дехлорирвоания фторхлоруглеродов водородом и окисления фторолефинов кислородом. На основе α-Al2O3:
- созданы стабильные катализаторы дехлорирования водородом 1,2-дихлоргексафторпропана до гексафторпропилена (Ni/α-AI2O3), 1.2-дифтортетрахлорэтана до 1,2-дифтордихлорэтилена (α-AI2O3; Ni/α-AI2O3), 1,1,2-трифтортрихлорэтана до трифторхлорэтилена (Cu,BaO/α-AI2O3), позволяющие существенно увеличить скорость, а также селективность этих процессов (с 70-80 до 90-95 % и выше); исследованы кинетика и механизм дехлорирования фторхлоруглеродов водородом;
- разработан эффективный низкотемпературный катализатор окисления тетрафторэтилена кислородом (оксид кобальта/α-AI2O3) с направленным получением карбонилдифторида.
3. В результате исследования кинетических закономерностей термических газофазных реакций полифторированных органических соединений установлено, что дегидратированный активированный уголь является эффективным агрессивостойким катализатором нуклеофильного типа и может быть использован даже в немодифицированном виде, в частности, для катализа изомеризации гексафторпропиленоксида, термодеструкции олигомеров карбонилдифторида и перфторированных полипероксидов, гидрофторирования и гидролиза префторизобутилена, а также его взаимодействия с 1,1,5-тригидроперфторпентанолом-1, разложения ω-хлорперфторалкилфторсульфатов и селективного гидролиза 1,1,1-трифтортрихлорэтана в смеси с 1,1,2-трифтортрихлорэтаном.
4. На основе разработанных агрессивостойких катализаторов термических газофазных превращений полифторированных органических соединений внедрены в производство:
- технология перфтордиметилперфторциклогексана – эффективной диэлектрической жидкости для авиационной техники;
- технология тонкой очистки основного компонента разрабатываемого искусственного кровезаменителя – перфтордекалина;
- технология очистки перфторполиоксаметиленацетилфторидов, позволившая полностью ликвидировать сброс этих токсичных фторангидридов и получить на их основе новое поколение термоморозоагрессивостойких фторкаучуков с уникальными свойствами.
Внедрены в опытно-промышленном масштабе:
- безотходная технология тонкой очистки фторуглеродных жидкостей и смазок с получением термохемостойких жидкостей Б-1, М-1 и смазок УПИ, КС и КСТ;
- технология регенерации отработанных фторуглеродных смазок и жидкостей;
- технология новой эффективной диэлектрической жидкости – 2-перфторметил-4-оксаперфторнонана.
Выданы исходные данные на проектирование:
- производства пентафторхлорэтана (хладона -115), с ликвидацией выброса и утилизацией октафторциклобутана – отхода производства тетрафторэтилена;
- технологии трифторметилгипофторида – реагента и инициатора для синтеза целого ряда новых практически важных кислородсодержащих фторорганических соединений;
- технологии гексафторпропилена, позволяющей ликвидировать выброс и, соответственно, утилизировать 1,2-дихлоргексафторпропан – отход в производстве тетрафторэтилена;
- технологии 1,2-дифтордихлорэтилена, исключающей образование и сброс токсичного хлорида цинка;
- технологии карбонилдифторида – основы для производства целого ряда кислородсодержащих фторорганических соединений;
- безотходной технологии очистки от перфторизобутилена продуктов пиролиза тетрафторэтилена (гидрофторированием) и кубовых продуктов в производстве гексафторпропилена (гидролизом);
- опытно-промышленной установки по селективной изомеризации гексафторпропиленоксида до перфторпропионилфторида – основы для производства эффективного инициатора сополимеризации фторолефинов.
Список литературы
2. Теддер Д.М. Фторирование органических соединений элементарным фтором. В кн."Успехи химии фтора", т. I-II -М.-Л. : Химия, 1964, с. 380-423.
3. Оркин В.Л., Чайкин A.M. Определение констант скорости образования атомов в реакциях молекулярного фтора с окисью азота, этиленом и тетрафторэтиленом. Кинетикаикатализ, 1982,т.23,в.З,с.529-533.
4. Anson P.O., Fredricks P.S., Tedder J.M. Free-radical substitutionin aliphatic compounds. Part 1. Halogenation of n-butahe and isobutane in the gas phase. J.Chem.Soc, 1959, March, p.918-922.
5. Fredricks P.S., Tedder. J.M. Free-radical substitution in aliphatic compounds. Part 11. Halogenation of the n-butylhalides. -J.Chem.Soc, 1960, Jan., p. 144-150.
6. Purington S.T., Kagen B.S., Patric T.B. The application of elemental fluorine in organic synthesis. Chem.Rev., 1986, № 86, p.997-1018.
7. Aikman R.E., Lagow E.J. Syhthesis of tetra-cis-(perfluorocyclohexyl)-methane and bis-(perfluorocyclohexyl)-methane by direct fluorination. -J.Org.Chem., 1982, v.47, № 14, p.2789-2790.
8. Еременко Л.Т., Орешко Г.В. Фторирование элементарным фтором лабильных полинитросоединений – аддуктов реакции Михаэля. -Изв. АН СССР, сер. Химия, 1969, № 2, с.479.
9. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская Й.П., Карпова Н.Д. Синтез перфторнитроэтанов. -Изв. АН СССР. Сер.хим., 1968, № 2, с.429-430.
10. Патент США 4523039. Способ получения перфорированных простых эфиров. /Лагов Р.Дж., Герхарт Дж.Е. -Опубл. 11.06.85, РЖ Химия, 1986, 7Н36.
11. Патент США 3242218. Способ получения фторуглеродных полиэфиров. /Миллер В.Т. -Опубл.22.03.66,РЖХимия, 1967, 12C232.
12. Des Martean D.D, Fluoroperoxytrifluoromethane CF300F. Preparation from trifluoromethyl hydroperoxide and fluorine in the presence of cesium fluoride. -Inorg Chem., 1972, v.11, № 1, p.193-195.
13. Merritt R.F., Johnson F.A. Direct fluorination. Addition of fluorine to indenes and.acenaphthylenes, -J.Org.Chem., 1966, v.31. № 6, p.1859-1863.
14. Merritt R.F, Johnson F.A. Direct fluorination of steroidal olefines to cis-vicinal difluorides. –J.Am.Chem.Soc., 1966, v.88, № 8, p.1822-1823.
15. Патент США.3487093. Фторированные олефины. Меррит Р.Ф. –Опубл.30.12.69,РЖХимия, 1970, 23Н29.
16. Grakauskas V. Direct liquid phase fluorination of halogenated aromatic compounds. -J.Org.Chem., 1969, v.34, № 10, p.2835-39.
17. Brooke G.M., Chambers B.D., Heyes J., Musgrave W.K.R. Direct preparation and some reactions ofсhlorofluorobenzenes. -J.Chem.Soc., 1964, Febr., p.729-735.
18. Merritt R.F, The polar addition of molecular fluorine to acetylenes. –J.Org.Chem., 1967, v.32, № 12, p.4124-4126.
19. Шеппард У., Шартс К. Органическая химия фтора. -
М.: Мир, 1972, с.52, 89, 112.
20. Миллер В., Эренфельд Дж, Фелан Дж., Пробер М., Рид Ш, Фторирование полностью галоидированных олефинов.«Химияфтора»,№ 2, -М.:ИЛ, 1950,с.228-240.
21. Miller W.T. My early days in fluorine chemistry. –J.Fluor.Chem., 1981, v.18, p.305-321.
22. Miller W.T., Stoffer J.O., Fuller G., Currie A.C. The mechanism of fluorination. IV. The effect of temperature and of fluorine concentration on reaction. A new fluorination apparatus. –
J.Am.Chem.Soc., 1964, v.86, № 1, p.51–56.
23. Исикава Нобуо, Китацумэ Томоя. Новые методы фторирования ароматических соединений. –Юкки госэй качаку кёкайси., 1976, т.34, № 3, с.173-178, РЖ Химия, 1976, 23Ж360.
24. Патент Японии 55-18695. Способ фторирования./ Маруо Кэйити, Мисаки Сусуму. –Опубл. 21.05.80, РЖ Химия, 1981, 5
Н122.
25.SiripL.A.,LagowR.J.Directfluorinationof 2,2,4,4-tetramethylpentane.Sterically protected residual protons. –J.Org.Chem., 1977, v.42, № 21, p.3437-3438.
26. Kowanko N., Branthaver J.F., Sugihara J.M. Direct liquid-phase fluorination of petroleus. –Fuel, 1978, v.57, № 12, p.769-775.
27. Патент США 40004996. Фторирование органических соединений./ Коллониц Дж. –Опубл. 25.01.77, РЖ Химия, 1977, 23Н57.
28. Патент Великобритании 1077065. Фторирование нитросоединений./ Гракаускас В., Хэмел Э.Э. –Опубл. 26.07.67, РЖ Химия, 1975, 16Н84.
29. Merritt R.F. Direct fluorination of 1,1-diphenylethylene. –J.Org.Chem., 1966, v.31, № 11, p.3871-3873.
30. Merritt R.F. The polar fluorination of propenylbenzene. –J.Am.Chem.Soc., 1967, v.89, № 3, p.609-612.
31. Merritt R.F., Johnson F.A. Low-temperature fluorination of Schiff bases. -J.Org.Chem., 1967, v.32, № 2, p.416-419.
32. Misaki S. Direct fluorination of phenol and cresols. –
J.Fluor.Chem., 1981, v.17, № 2, p.159-171.
33. Maxwell A.F., Detoro F.E., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The jet fluorination of certain aliphatic hydrocarbons as oriented and controlled by operation conditions. -J.Am.Chem.Soc., 1960, v.82, № 22, p.5827-5830.
34. Attaway J.A., Groth R.H., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIII. The fluorination of some amides, nitriles and of methylthiocyanate. -J.Am.Chem.Soc., 1959, v.81, № 14, p.3599-3603.
35. Robson P., Mc Longhlin V.C.R., Hynes J.B., Bigelow L.A. The action of elementary fluorine upon organic compounds. XXIV. The jet fluorination of hydrogen cyanide, cyanogen, methylamine and ethylenediamine. Pyrolysis and fluorinolysis of selected products. -J.Am.Chem.Soc., 1961, v.83, № 24, p.5010-5015.
36. Boffenberg K. Substitution von benzol durch elementares Fluor in der Gasphase. –Chem.Ztg.,1972, v.96, № 2, p.84-92.
37. Патент Японии 58-41829. Получение октафторпропана./ Фукуи Сиро, Йонэда Хадзимэ. –Опубл.11.03.83,РЖХимия, 1984, 5Н11.
38. Hayes L.J., Dixon D.D. Direct fluorination of polyester and related compounds. –J.Fluor.Chem., 1977, v.10, № 1, p.1-16.
39. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropolyethers by direct fluorination: a novel preparation for perfluoro (polypropylene oxide) ethers and perfluoro (polymethylene oxide) ethers. –J.Chem.Soc., 1981, part 1, № 5, p.1321-1328.
40. Патент США 4113772. Метод получения олигомеров перфторэфиров с концевыми карбоксильными группами. / Лагов Р.Дж. –Опубл.12.09.78,РЖХимия, 1979, 15Н14.
41. Gerhardt G.E., Lagow R.J. Synthesis of perfluoropoly (ethylene glycol) ethers by direct fluorination. –J.Org.Chem., 1978, v.43, № 23, p.4505-4509.
42. Adcock J.L., Znoue Shoji, Lagow R.J. Simultaneous fluorination and functionalization of hydrocarbon polymers. –J.Amer.Chem., 1978, v.100, № 6, p.1948-1950.
43. Lagow R.J., Margrave J.L. The controlled reaction of hydrocarbopolymers with elemental fluorine. –J.Polym.Sci.: Polym.Lett.Ed., 1974, v.12, № 4, p.177-184.
44. Pat. 3775489 (USA). Process for fluorination of aromatic and polynuclear hydrocarbon compounds and fluorocarbons produced thereby. Margrave J.L., Lagow R.J. –Publ. 27.11.73.
45. Lagow R.J., Maraschin N.J. Direct fluorination of cyclic and bicyclic hydrocarbons. –7th Int.Symp.Fluorine Chem., Santa Cruz, Calif., 1973, s.l., s.a., p.1-25.
46. Gerhardt G.E., Dumitru E.T., Lagow R.J. Synthesis of hightbranched perfluoroethers by direct fluorination, promising new materials based on the hexafluoroacetone –ethylene copolymer. –J.Polym.Sci., 1980, v.18, № 1, p.157-169.
47. Robertson G., Liu E.K.S., Lagow R.J. Synthesis of perfluoroadamantane compounds by direct fluorination. –J.Org.Chem., 1978, v.43, № 26, p.4981-4983.
48. Lagow R.J. Large scale synthesis of organofluorine compounds using elemental fluorine; a third Simons cell. –Int.Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.2.
49. Adcock J.L., Lagow R.J. The synthesis of the fluorinated ethers “perfluoroglyme” and “perfluorodiglyme” by direct fluorination. –J.Org.Chem., 1973, v.38, № 20, p.3617-3618.
50. Koshar R.J., Hausted D.R., Meiklejohn R.A. Organic fluoronitrogens. V. Bis(difluoroamino)difluoromethane. –
J.Org.Chem., 1966, v.31, № 12, p.4232-4234.
51. Koshar R.J., Hausted D.R., Wright C.D. Organic fluoronitrogens. VII. Tris(difluoroamino)fluoromethane and related compounds. –J.Org.Chem., 1967, v.32, № 12, p.3859-3864.
52. Патент США 3981783. Процесс электрохимического фторирования с дополнительной подачей водорода и увеличением выхода по току. / Чайлдс В.В. –Опубл.21.09.76, РЖ Химия, 1977, 14Л237.
53. Патент Германии 2106870. Способ электрохимического фторирования органических соединений./Восс П., Нидерпрум Х., Кауль Г., Трепп Р., -Опубл. 24.02.77, РЖ Химия, 1977, 24Н16.
54. Патент Германии 2302132, 1976. Способ получения разветвлённых перфторалканов. /Беннингер С. -Опубл. 23.12.76, РЖ Химия, 1977, 24Л202.
55. Патент США 4035250. Способ получения перфторгептана. /Вальтерс Х.С., Чайдс В.В. -Опубл.12.07.77, РЖ Химия, 1978, 6Н22.
56. Патент Японии 53-18488. Способ получения перфторциклоалканов. /Сато Дайсукэ, Ямамути Коити, Мурасима Рёширо. -Опубл. 15.06.78, РЖ Химия, 1979, 9Н121.
57. Патент США 3662009. Получение ненасыщенных фторсодержащих соединений. /Хадчинсон В.М. -Опубл. 09.05.72, РЖ Химия, 1973, 9Н12.
58. Гольдштейн Б.В., Серушкин И.Л., Никонорова Н.И. О роли фторида никеля в реакциях электрохимических соединений. –Тр. ГИПХ, № 39, часть 1, инв. № Т-1863., Л., 1975, с.43-47.
59. Фаулер Р.Д., Берфорд Н.Б., Гамильтон Дж.М., Свит Р.Дж., Уэбер К.Е., Каспер Дж.С. Лайтайн Н. Синтез фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.91-113.
60. Беннер Р.Дж., Беннинг А.Ф., Доунинг Ф.Б., Ирвин С.Ф., Джонсон К.С., Линч А.Л., Пармели Х.М., Уирт У.У. Фторуглероды, полученные фторированием углеводородов трехфтористым кобальтом. В кн. «Химия фтора», сб. № 1 –М.: ИЛ, 1950, с.114-128.
61. Фаулер Р.Д., Андерсон Х.С., Гамильтон Дж.Н., Берфорд Н.Б., Спагетти А., Битердих С.Б., Лайтайн Н. Фториды металлов, применяемые в синтезе фторуглеродов. В кн. «Химия фтора», сб. № 1 –М.:ИЛ, 1950, с.143-153.
62. Патент Японии 60-81134. Получение октафторпропана. /Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 09.05.85, РЖ Химия, 1986, 12Н25.
63. Патент Японии 60-10933. Получение гексафторэтана./ Катамура Коити, Кагэяма Ютака, Накаяма Хидэтоси. –Опубл. 15.06.85, РЖ Химия, 1986, 11Н21.
64.PakV.,PekaJ.,CermakV.,SykoraF.,PetrzilaV.Poloprovoznizarizeniprovyrobuperfluororganickychlatek.
–Chem.Prum., 1978,v.28, № 9,p.467-470.
65. Патент Франции 2381732. Усовершенствованный способ перфторирования циклических углеводородов. / Мур Р.Е. –
Опубл. 27.10.78, Изобретения в СССР и за рубежом, 1979, в.55, № 5, с.67.
66. Патент США 4143079. Способ получения перфтор-1-метил-4-изопропилциклогексана из пинена. / Мур Р.Е. –Опубл. 06.03.79, Изобретения в СССР и за рубежом, 1979, в.55, No 21, с.20.
67. Патент Великобритании 1597914. Способ перфторирования циклических углеводородов. / Сантэч инкорп. –Опубл. 16.09.81, Изобретения в СССР и за рубежом, 1982, в.57, № 12, с.64.
68. Burdon J. The exhaustive fluorination of aliphatic compounds.
–Int. Symp. “Cent. ...”, Paris, 1936, p.9.
69. Moore R. E., Driscoll G. Perfluorination of bicyclic and tricyclic hydrocarbons. –IV-th Winter Fluorine Conf., Abstr, Daytona Bearen, 1979, p.8.
70. Асович В.С., Прокудин И.П. Особенности фторирования органических веществ четырехфтористым церием. –Тр. ГИПХ, № 40, инв. № Т-2117, Л., 1976, с.25-31.
71. Исикава Н., Кобаяси Ё. Фтор. Химия и применение. –
М.: Мир, 1982, с.91.
72. Кэйджи Дж..Х., Гроссе А.В., Барбер Е.Дж., Бэрджэр Л.Л., Шелдон З.Д. Получение фторуглеродов каталитическим фторированием углеводородов. В кн. «Химия фтора», сб. № 1 –М.: ИЛ., 1950, с.129-135.
73. Бигелоу Л.А. Действие элементарного фтора на органические соединения. В кн. «Фтор и его соединения (под ред. Дж. Саймсона), т.1 –М.: ИЛ., 1953, с.314-335.
74. Musgrave W. K. R., Smith F. Organic Fluorides. PartІІ. The effect of metals on the fluorunationof hydrocarbons. –J. Chem. Soc., 1949, p.3026-3028.
75. Брик Т. Дж. Фторуглероды; их свойства и производство во время войны. В кн. «Фтор и его соединения» (под ред. СаймонсаДж.),т.І –М.:ИЛ, 1953,с.355-388.
76. Maraschin N. J., Catsikis B. D., Davis L. H., Larvinen J., Lagov R. J. Synthesis of structurally unusual fluorocarbons by direct fluorocarbon. –J.Am. Chem. Soc., 1975, v. 97, № 3, p.513-517.
77. Патент США 4113453. Аппарат для низкотемпературного прямого фторирования. / Лагов Р.Дж, Адкок И.Л., Марашин Н.И. –Опубл. 12.09.78, РЖ Химия, 1979, 19И130.
78. Патент США 4281119. Аппарат для прямого фторирования с контролируемым охлаждением по зонам. / Лагов Р.Дж., Адкок И.Л., Марашин Н.И. –Опубл. 28.07.81, РЖ Химия, 1982, 9Н225.
79. Рахимов А.И., Химия и технология фторорганических соединений. –М.: Химия, 1986, с.9-10.
80. Schmeisser M., Ehlers K. P., Sartori P. Dichlorhexafluorpropan durch fluorierung von 1,2-dichlorpropan. –Angew. Chem., 1967, v.79,№ 13, p.622.
81. Margrave W. K. R., Smith F. Organic Fluorides. PartІ. Fluorination of hydrocarbons. –J. Chem. Sоc., November, 1949, p.3021-3026.
82. Гроссе А.В., Кэйди Дж. Х. Свойства фторуглеродов. В кн. «Химия фтора: сб. № 1 –М.: ИЛ, 1950, с.35-64.
83. Patent GB 1281822. Improved Fluorination Process. / Kingdom R. J., Bond G. D. –Publ. 19.07.72.
84. Hill M. Process and market development of fluorocarbon fluids. –Chem.аnd Ind., 1975, № 3, p.118-121.
85. Патент Франции 2028457 (В). Способ фторирования. / Империал Смелтинг Корп. –Опубл. 15.01.70, Офф. Бюлл. Франции, Химия и металлургия, 1970, № 45-48, ч. І, с.44.
86. Патент США 4220606. Способ получения перфторпроизводных из цикличеких углеводородов. / Мурр Р.Е. –Опубл.02.09.80,РЖХимия, 1981, 9Н105.
87. Patent US 3480667. Method of producing fluorinated compounds. / Siegart W. R., Blackley W. D. –Publ. 25.11.69.
88. Патент США 4330475. Аэрозольный способ прямого фторирования и устройство для этой цели. / Адкок Д.Л., Рэнк Е.Б. –Опубл.18.05.82,РЖХимия, 1983,ІІН76.
89. Ruff J. K. The catalytic fluorination of perfluorocarbon nitriles and imines. –J. Org. Chem., 1967, v.32,№ 5, p.1675-1677.
90. Lusting M., Ruff J. K. Fluorination of some perfluoro alkyliminosulfurdifluorides. –Inorg. Chem., 1965,v.4, № 10,p.1444-1446.
91. Фокин А.В., Столяров В.П., Радченко В.П. Синтезполифторамино-и α-фторнитросоединений на основе реакций газообразного фтора. –Изв. АН СССР, сер. хим., 1982, № 8, с.1853-1861.
92. Фокин А.В., Галахов В.С., Узун А.Т. и др. Реакция фтора с солями щелочных металлов динитроацетонитрила в присутствии фторидов калия или кальция. –Изв. АН СССР, сер. хим., 1974, № 2, с.456-458.
93. Фокин А.В., Узун А.Т., Столяров В.П. Бис (дифторамино) фторацетальдегид –новый представитель α, α-бис (дифторамино) альдегидов. –Изв. АН СССР, Сер. хим., 1982, № 6, с.1438.
94. Еременко Л.Т., Нацибуллин Ф.Я., Боровинская И.П., Карпова Н.Д. Синтез перфторнитроэтанов. –Изв. АНСССР,Сер.хим., 1968, № 3,с.431-432.
95. Sekiya A., Des Martean D. D. Synthesis of 1,1-bis (fluoroxy) –perhaloalkanes by reaction of fluorinated carboxylic acids with fluorine in the presence of cesium fluoride. –Inorg. Chem., 1980, v. 19,№ 5, p.1328-1330.
96. Lu S., Des Martean D. D. Direct synthesis of fluorinated peroxides. 7. Perfluoro-tret-butyl-fluoroformyl peroxide. –Inorg. Chem., 1978, v. 17, № 2, p.304-306.
97. Schack C. J. A new synthesis of difluoraminotrifluoromethane. –J. Fluor. Chem., 1981,v. 18,no 4,p.583-586.
98. Патент Германии 2712732. Способ получения октафторпропана. /Халац С.П. –Опубл. 28.09.75, РЖ Химия, 1979, 14Н14.
99. Патент США 4158023. Способ получения октафторпропана. / Халац С.П. –Опубл. 12.06.79. Изобретения в СССР и за рубежом, 1979, в. 55, № 24, с.121.
100. Патент Великобритании 1568020.Способ получения октафторпропана. / Халац С.П. –Опубл. 21.05.80. Изобретения в СССР и за рубежом, 1981, в. 55, № 3, с.54.
101. Патент США 4377715. Получение перфторпропана. /Нучка Х.Р., Хино И.Б., Эйбэк Р.Е., Робинсон Н.А. –Опубл. 22.03.83, Изобретения в СССР и за рубежом, 1983, в.57, № 23, с.88.
102. Европейский патент 0031519. Способ фторирования органических соединений элементарным фтором. / Эллайд Кэмикэл Ко. –Опубл. 08.07.81, Изобретения в СССР и за рубежом, 1983, в.57, No 1, с.20.
103. Европейский патент 0032210. Способ фторирования органических соединений фтором в трубчатом реакторе из пористого металла в присутствии перфторированного разбавителя. / Эллайд Кэмикл Ко. –Опубл. 22.07.81, Изобретения в СССР и за рубежом, 1983, в.57, № 2, с.27.
104. Патент США 4513154. Способ проведения последовательно-конкурирующих газофазных реакций. / Курц Б.Е. –Опубл. 23.04.85, РЖ Химия, 1986, 4Н25.
105. Патент США 2831035. Производство фторированных углеродов. / Тучковский Э.А., Вульф К. –Опубл. 15.04.58, РЖ Химия, 1960, 85740.
106. Патент США 3709800. Получение перфторированных углеводородов./ Фокс Х.М. –Опубл. 09.01.73, РЖ Химия, 1973, 22Н23.
107. Патент Японии 5608. Фторсодержащие галоидуглеводороды. / Осиба Такаси. –Опубл. 22.03.65, РЖ Химия, 1968, 5Н35.
108. Патент США 2681267. Процесс улучшения каталитических свойств фтористого алюминия и продуктов из него. / Калфи Дж. Д., Миллер Ч.Б. –Опубл.15.06.54,РЖХимия, 1956, № 14, 45553.
109. Hohorst F. A., Shreeve J. M. Bis-(fluoroxy)-difluoromethane,CF2(OF)2. –J. Am. Chem. Soc., 1967, v.89, № 8, p.1809-1819.
110. Walker N., Des Martean D. D. Direct Synthesis of fluorocarbon peroxides.ІІІ. The addition of chloroperoxytrifluoromethane to olefins. –J. Am. Chem.Soc., 1975, v.97,№ 1, p.13-17.
111. Патент США 4499024. Непрерывный способ получения бис (фторокси) дифторметана. / Филолт М.Ж. –Опубл.12.02.85,РЖХимия, 1985, 20Н14.
112. Lusting M. A., Pitochelli A. R., Ruff J. K. The catalytic addition of fluorine to a carbonyl group. Preparation of fluoroxy compounds. –J. Am. Chem. Soc., 1967, v. 89, № 12, p.2841-2843.
113. Ruff J. K., Pitochelli A. R., Lusting M. A. A simple synthesis of fluoroxyperfluoroalkyl compounds. -J. Am. Chem. Soc., 1966, v.88,№ 19, p.4531-4532.
114. Kennedy R. C., Cady G. H. Reaction of carbonyl fluoride with fluorine in the presence of various fluorides as catalysts. –J. Fluor. Chem., 1973,v.3, № 1, p.141-154.
115. Мухаметшин Ф. М. Успехи химии фторорганических гипогалогенитов и родственных соединений. –Успехи химии, 1980, т. 49, № 7, с.1260-1288.
116. Мухаметшин Ф.М. Гипофториты и их применение в органическом синтезе. –В кн. «Новые фторирующие реагенты в органическом синтезе». –Новосибирск: Наука, 1987, с.140-196.
117. Cady G. H. Proposed mechanisms of catalysis of fluorination of CF2O. –Anales. Asoc. Quim. Argentina, 1971, v.59, p.3-4, p.125-131.
118. Patent US 3230264. Reaction of carbonyl fluoride with fluorine. –Roger S. P., Cady G.H. –Publ. 18.01.66.
119. Wechsberg M., Cady G.H. Comparative studies of catalytic fluorination of carbon monoxide with elementary fluorine. –J. Am. Chem. Soc., 1969, v.91, p.4432-4435.
120. Kellog K. B., Cady G.H. Trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1948, v.70,№ 12, p.3986-3988.
121. Gervasi J.A., Brown M., Bigelow L. A. The action of elemental fluorine upon organic compounds. XX. The fluorination of mono-, di-and trimethylamine, ethylenediamine and ethyleneimine. –J. Am. Chem. Soc., 1956, v.78, № 8, p.1679-1682.
122. Avonda F. P., Gervasi J. A., Bigelow L.A. The action of elemental fluorine upon organic compounds. XXІ. The fluorination of malononitrile and dimethylformamide. –J. Am. Chem. Soc., 1956, v.78,№ 12, p.2798-2800.
123. Тойтельбойм М.А., Шойхет А.А., Каплунов М.Г., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. –Кинетика и катализ, 1981, т. 22, в.2, с.298.
124. Тойтельбойм М.А., Шойхет А.А., Веденеев В.И. Энергетические разветвления цепей в реакциях трифторметилгипофторита с галоидметанами. 2. Механизм отрицательного взаимодействия цепей. –Кинетика и катализ,
1981, т.22, в.3, с.564-568.
125. Веденеев В.И., Парийская А.В. Механизм фторирования метана и его фторпроизводных. І. Сравнение скоростей фторирования метана, фторметана, дифторметана и трифторметана. –Кинетика и катализ, 1971, т.12, в.1, с.21-26.
126. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его производных. 2. Дифторметан –Кинетика и катализ, 1971, т.12, в.2, с.293-298.
127. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 3. Фтористый метил. –
Кинетика и катализ, 1971, т.12, в.3, с.543-548.
128. Парийская А.В., Веденеев В.И. Механизм фторирования метана и его фторпроизводных. 4. Метан. –Кинетика и атализ, 1971, т. 12, в.4, с. 839-842.
129. Надточенко В.А., Федотов Н.Б., Веденеев В.И., Саркисов О.М. О реакции колебательно возбужденной молекулы СН3F с фтором. –Докл. АН СССР, 1978, т.238, № 6, с.1391-1394.
130. Парийская А.В., Веденеев В.И. О природе задержек самовоспламенения в системе СН3F + F2 + O2 + Не. –Кинетика и катализ, т.14, в.6, с.1365-1369.
131. Веденеев В.И., Тейтельбойм М.А., Шойхет А.А. Фотохимическое фторирование фтороформа в присутствии кислорода. –Изв. АН СССР, 1976, № 9, с.1968-1970.
132. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Химическая активация солекулы СHFCl2 в реакции СНCl2+ F2→ СНСl2F + F . –Кинетика и катализ, т.12, в.2, с.269-275.
133. Медведев Б.А., Тейтельбойм М.А., Шилов А.Е. Механизм газофазной реакции хлорфторметана с молекулярным фтором. –Кинетика и катализ, 1971, т.12, в.3, с.49-751.
134. Веденеев В.И., Медведев Б.А., Тейтельбойм М.А. Газофазное фторирование дифторметана при повышенных давлениях инертного газа. –Кинетика и катализ, т.13, в.1, с.50-53.
135. Обвивальнева А.А., Федотов В.Г. Возбужденные молекулы в реакции фтора с ацетоном (фторацетоны; синтез; механизм). –Кинетика и катализ, 1981, т.22, в.5, с.1095-1099.
136. Капралова Г.А., Марголина Е.М., Русин Л.Ю., Чайкин А.М., Шилов А.Е. Роль колебательно-возбужденных молекул в реакциях фторирования молекулярным фтором. –В сб. «5 Междунар. симпозиум по химии фтора», Тезисы докл., М., : Наука, 1969, с.102-104.
137. Кнунянц И.Л, Гамбарян Н.П., Рохлин Е.М. Карбены. (Соединения двухвалентного углерода, промежуточно образующиеся в органических реакциях). –Успехи химии, 1958, т.27, в.12, с.1361-1436.
138. Мухаметшин Ф.М., Жирнов О.М., Айнагос Н.А., Петров Ю.И., Ляпунов М.И. О некоторых особенностях гипофторитного метода получения перфторметилвинилового эфира. Сообщ. 2. Синтез трифторметилгипофторита. –Тр. ГИПХ, № 69, инв. № Т-2769, Л., 1980, с.73-78.
139. Мухаметшин Ф.М., Поврозник С.В., Жирнов О.М. Исследование кинетики и механизма каталитической реакции образования трифторметилгипофторита. –Тр. ГИПХ, № 103, инв. № Т-2981, Л., 1983, с.75-80.
140. Авторское свидетельство СССР 1141707. Способ получения 1,1-дифтор-1-хлорэтана или 1,1,2,2-тетрафтордихлорэтана. /Захаров В.Ю., Голдинов А.Л., Боровнев Л. М., Голубев А.Н., Калашникова Н.А., Захарова О.М., Любимова Л.А.
141. Капралова Г.А., Бубен С.Н., Чайкин А.М. Кинетика и механизм реакции фторирования окиси углерода. –Кинетика и катализ, 1975, т.16, в.3, с.591-595.
142. Поврозник С.В. Разработка способа и технологии получения фторокситрифторметана реакциями элементарного фтора с карбонилсодержащими соединениями. Дисс. канд. техн. наук, 1987, ПФ НПО ГИПХ, 161с.
143. Краснов К.С. Молекулы и химическая связь. –М.: Высш. школа, 1977, с.117.
144. Безмелицын В.Н., Легасов В.А., Чайванов Б.Б. Синтез дифторида криптона с применением термической генерации потоков атомарного фтора. –Докл. АН СССР. Сер. хим., 1977, т.235, № 1, с.96-98.
145. Безмелицын В.Н., Легасов В.А., Спирин С.Н., Чайванов Б.Б. Кинетика каталитической атомизации фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, № 5, с.1153-1157.
146. Палкина Л.А., Спирин С.Н., Тищенко П.П. процессы с участием атомарного фтора. –Докл. АН СССР. Сер.физ.химия, 1982, т.262, с.1428-1433.
147. Безмелицын В.Н., Васильев А.А., Синянский В.Ф., Чайванов Б.Б. Кинетика термокаталитической диссоциации фтора на поверхности никеля. –
Тезисы докладов на 8 Всесоюзн. симп. по химии неорг. фторидов, 1987, М.: Наука, с.59.
148. Никоноров Ю.И. Фториды никеля. Их каталитическая и окислительная активность. –Тезисы докладов на 5 Всесоюзн. симп. по химии неорг. фторидов, 1978, М.: Наука, с.205.
149. Рысс И.Г. Химия фтора и его неорганических соединений. –М.: Госхимиздат, 1956, с.576-579.
150. Redwood M.E., Willis C. J. Fully fluorinated alkoxides. PartІ. Trifluoromethoxides of alkali metals. –Canad. J.Chem., 1965,v.43,p.1893-1898.
151. Губанов В.А., Веретенников Н.В., Тройчанская П.Е., Долгопольский И.М. Синтез перфторалкоксидов металлов І группы и термографическое изучение их свойств. –Ж. Орг. хим., 1975, т.ІІ, № 2, с.322-325.
152. Levy J. B., Kennedy R. C. Bis (trifluoromethyl) peroxide.І. Thermodynamics of the equilibrium with carbonyl fluoride and trifluoromethyl hypofluorite. –J. Am. Chem. Soc., 1972, v.94, no 10, p.3302-3305.
153.Патент Японии 35888.Способполучениятрифторметилгипофторита. /НагасэСюндзи,АбэТакаси,БабаХадзимэ,ОхираКадзуо. –Опубл.09.09.72, РЖХимия, 1973, 14Н120.
154. Патент США 3687825. Способ получения трифторметилгипофторита. Нагасэ Сюндзи, Абэ Такаси, Баба Надзимэ. –Опубл. 29.08.72, РЖ Химия, 1973, 21Н26.
155. Патент США 2983764. Способ получения фторуглеродных соединений./ Кнак Д.Ф. –Опубл. 9.05.61, РЖ Химия, 1962, 14Л53.
156. Патент Японии 56-16429. Олигомеризация гексафторпропилена в газовой фазе. / Хосака Йоносукэ, Хигасидзука Такэси. –Опубл. 17.02.81, РЖ Химия, 1982, 3Н10.
157. Патент Японии 50-45818. Получение олигомеров гексафторпропилена. Кадзава Масахиро, Комацу Тадааки, Мацуока Кимиаки. –Опубл. 08.06.81, РЖ Химия, 1982, 9НІІ.
158. Патент США 4296265. Способ получения олигомеров гексафторпропилена. / Хосака Йоносукэ, Тозука Такаси. –Опубл. 20.10.81, РЖ Химия, 1982, І3НІІ.
159. Патент Германии 3027229. Способ получения олигомеров гексафторпропилена. / Осака Ю., Тозука Т. –Опубл. 07.02.81, Изобретения в СССР и за рубежом, 1981, В.55, № 12, с.30.
160. Патент Бельгии 2055824А. Способ получения олигомеров гексафторпропилена. /Осака И., Тозука Т. –Опубл. 18.07.80, Сборник рефератов НИОКР, обзоров, переводов и деп. рукописей. ДСП. Химия и химическая технология, М., 1986, № 5, с.14.
161. Патент Германии 2616733. Способ получения ди
-и тримеров гексафторпропилена. / Окава М., Комацу Т., Мацуока К. –Опубл. 20.03.80, Изобретения в СССР и за рубежом, 1980, № 15, с.13.
162. Патент Японии 57-2697. Способ получения олигомеров гексафторпропена. / Нэосу К.К. –Опубл. 18.01.82, Изобретения в СССР и за рубежом, 1982, в.57, № 14, с.113.
163. Scherer K. V., Ono T., Jamanouchi K., FernandezR., Henderson P.B. F-2,4-Dimethyl-3-ethyl-3-pentyl and F-2,4-dimethyl-3-isopropyl-3pentyl: stable tert-perfluoroalkyl radicals prepared by addition of fluorine or trifluoromethyl to a perfluoroalkene. –J. Am. Chem. Soc., 1985, v.107, № 3, p.718-719.
164. Scherer K. V., Fernandez R., Henderson P.B. Long lived perfluoroalkyl radicals: preparation by direct fluorination and alternate routes, properties and rates andmechanisms of disappearance. –Int. Symp. “Centenary of the discovery of fluorine”, Abstr., Paris, 1986, p.1.
165. Патент Германии 2332097. Способ получения перфторнонанов. /Халац С.П. –Опубл.16.01. 75,РЖ Химия, 1975, 18Н25.
166. Патент Германии 2332088. Способ получения перфтор-2-метилпентана./ Халац С.П. –Опубл. 16.07.81, Изобретения в СССР и зарубежом, 1981, в.55, № 23, с.21.
167. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Кирюхин Д.П. Образование стабильных радикалов из фторорганических соединений. –Изв. АН СССР. Сер. хим., 1983, No 6, с.1225-1228.
168. Аллаяров С.Р., Баркалов И.М., Гольданский В.И., Демидов С.В., Кирюхин Д.П., Михайлов А.И. Стабильные радикалы –растворенные перфторалкилы. –Изв. АН СССР. Сер. хим., 1983, № 6, с.1448-1449.
169. Аллаяров С.Р., Михайлов А.И., Баркалов И.М. Новый стабильный перфторалкильный радикал. –Изв. АН СССР. Сер.хим., 1985, № 7, с.1667-1669.
170. Аллаяров С.Р., Баркалов И.М., Лебедов М.Ю., Логинова Н.Н., Михайлов А.И. Образование стабильных радикалов при радиолизе гексафторпропилена. –Изв. АН СССР. Сер. хим., 1986, № 2, с.462-463.
171. Аллаяров С.Р., Баркалов И.М., Гольданский В.И. Образование долгоживущих радикалов при фторировании жидких углеводородов. – Изв. АН СССР. Сер. хим., 1986, № 10, с.394.
172. Аллаяров С.Р., Сумина И.В., Баркалов И.М., Михайлов А.И. Образование стабильных радикалов при
фотолизе перфтор-2,4-диметил-3-этилпентена-2. –Изв. АН СССР, Сер. хим., 1986, № 10, с.2359-2361.
173. Krusic P. J., Scherer K. V. An electron spin resonance study of long lived fluoroalkyl radicals generated by photolysis of hindered perfluoroalkenes. Int. Symp. “Centenary of the discovery of fluorine”. Abstr., Paris, 1986, p.37.
174. Levy J.B., Kennedy R. C. Homolytic displacement reactions of carbon.І. The fluorine-perfluorocyclobutane reactions. –J.Am.Chem.Soc., 1974, v.96, № 15, p.4791-9795.
175. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я., Жукова В.А., Новикова М.Д. Новые аспекты прямого фторирования. Каталитическое газофазное фторирование фторолефинов; получение перфторуглеродов. –Отчет предприятия п/я А-1619, 1987г., инв. № 1288, 87с.
176. Матвеев С.А., Прокудин Л.П., Тимофеев Н.В. Использование трифторида кобальта для фторирования органических соединений по двойным связям. –Тр. ГИПХ, 1975, № 31, инв. № Т-1728, с.119-127.
177. Асович В.С., Прокудин И.П. Фторирование ацетилена фторидами кобальта и церия. Тр. ГИПХ, № 55, инв. № Т-2552, Л., 1978, с.21-24.
178. Асович В.С., Прокудин И.П., Максимов Б.Н., Степанов В.П. Изучение кинетических закономерностей фторирования ацетилена трехфтористым кобальтом. –Тр. ГИПХ, № 65, инв. № Т-2723, Л., 1979, с.42-47.
179. Котиков С.В., Прокудин И.П., Асович В.С. Изучение кинетических закономерностей фторирования гексафторпропилена трехфтористым кобальтом. –Тр. ГИПХ, № 101,инв. №Т-2986,Л.,ГИПХ, 1985,с.24-27.
180. Burdon J., Knights J.R., Parsons J. W., Tatlow J. C. The fluorination of ethane and ethene over potassium tetrafluorocobaltate (ІІІ) and cobalt trifluoride. –Tetrahedron, 1976, v.32, p.1041-1043.
181. Гольдинов А.Л., Боровнев Л.М., Широкова Н.С., Ковбасюк О.Г., Тишина В.В. Влияние технологических параметров синтеза и чистоты растворителя на свойства фторопласта-50. –Отчет предприятия п/я А-1619, 1987, инв. № 1231.
182. Патент Германии 2451493. Способ получения перфторированных эфиров./ Халац С.П., Клаг Ф. –Опубл.06.05.76,ИзобретениявСССРизарубежом,1982,в.57, № 22,с.5.
183. Patent US 3665041 (USA). Perfluorinated polyethers and process for their preparation. / Sianesi D., Fontanelli R., -Publ. 23.05.72.
184. Рябинин Н.А., Долубенов А.Е., Шматов Л.Е. Фторполиэфиры как антиадгезивы при прессовании перхлората нитрония. –Тр. ГИПХ, № 101, инв. № Т-2986, Л., 1985, с.83-87.
185. Авторское свидетельство СССР 1332758. Способ очистки гексафторпропилена, тетрафторэтилена и их семей от октафторизобутилена. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
186. Гольдинов А.Л., Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Разработка технологии получения 2
-перфторметил-4-оксаперфторнонана на основе перфторизобутилена и 1,1,5-тригидроперфтор пентанола. –Отчет предприятия п/я А-1619, 1987, инв. № 1229, 39с.
187. Авторское свидетельство СССР 1148285. 3-Гидропропилфторсульфат в качестве промежуточного продукта для синтеза 2,2,3,3-тетрафторпропионовой кислоты и способ ее получения. /Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
188. Заявка СССР 3966753. Способ получения 2-гидротетрафторэтилфторсульфата. /Фокин А.В., Рапкин А.И., Татаринов А.С. и др. Пол. реш. от 26.09.86.
189. Авторское свидетельство СССР 1233595. Способ получения ω-гидроперфторкарбоновых кислот (его варианты). / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
190. Технические требования к ПФДМЦГ для использования в радиоэлектронике. М.: ИНЭОС АН СССР. Рег. № 12111-10 ДСП от 18.01.85.
191. Исходные данные для разработки жидкости-диэлектрика. М.: ИНЭОС АН СССР. Прил. к вх. № 155 от 18.02.85.
192. Гольдинов А.Л., Захаров В.Ю., Байбаков П.Я. и др. Разработка технологии получения перфтордиметилциклогексана повышенной чистоты. –Отчет предприятия п/я А-1619, 1986, инв. № 1218, 24с.
193. Гольдинов А.Л., Масляков А.И., Захаров В.Ю. и др. О возможности резкого увеличения выпуска дефицитных фторуглеродных жидкостей и смазок. –Отчет предприятия п/я А-1619, 1986, № 1219, 13с.
194. Временный технологический регламент производства перфтордекалина.1981,инв. № 1115.
195. Furin G.G. Synthetic aspects of the fluorination of organic compounds. (Ed.) M.E. Vol’pin./Harwood Academic
Publishers GmbH: United Kingdom, Sov. Sci. Rev., B. Chem., 1991, V.16, Part 1,р.140.
196. Rozen S., Kol M. Olefin epoxidation using elemental fluorine. / J.Org.Chem., 1990. V.55,р.5155-5159.
197. Patent JP 02-131438, (1990). CO719/08. Preparation of hexafluoroethane. /Shimuzu M.C.A. 1990, V.I 13,97031.
198.PatentJP62-193946, (1989).CO719/08,CO17/04. Способ получения фторсодержащих этанов. /Фарука Я., Коно Х. РЖХ, 1990, 2Н 131.
199. Lagow R.J. Important new developments in direct fluorination technology. /Memorial symposium for Prof. Nobuo Ishikawa, Dec. 9-10, 1991, Nippon Kaiun Club, Japan.
200. Lin W.H., Lagow R.J. Synthesis of perfluorodicyclohexano-crown-6 ether. /J.Chem.Soc., Chem.Commun., 1991,р.13-14.
201. Clark W.D., Lin T.Y., Maleknia S.D., Lagow R.J. Synthesis of perfluorotetralkyl ortocarbonates using elemental fluorine. / J.Org.Chem., 1989. V.54.р.1990-1992.
202. Sievert A.C., Tong W.R., Nappa M.J. Preparation D’analogues fluores de la pheromone d’anthanomus grandis. / J.Fluorine Chem., 1991. V.51.р.397-405.
203. Tonelli C., Tortelli V. Photoinduced fluorination of hexafluoropropene trimers: synthesis of branched perfluoroalkanes. /J.Chem.Soc., Perkin Trans I, 1990, № 1,р.23-26.
204. Patent Application EP 344935, (1989). CO8G65/00. Preparation of prefluoroethers by photo-assisted fluorination. / Nappa M.J., Sievert A. C.A. 1990, V. 112, 216229.
205. Patent US 4960951, (1990). CO743/12. Novel perfluoroalkyl ethers by two-step fluorination of polyols./ Nappa M.J., Sievert A.C. Tong W.R. C.A. 1991, V. 111, 216229.
206. Sievert A.C., Tong W.R., Nappa M.J. Synthesis of perfluorinated ethers by an improved solution phase direct fluorination process. / J.Fluorine Chem. 1991.V. 53, р. 397-417.
207. Мизин Г.Г., Симаков Б.В., Шкультецкая Л.В., Молдавский Д.Д. Исследование способа получения перфторэтилциклогексена. / ЖПХ. 1991. т.64 ,в.6. с.293-1296.
208. Moldavsky D.D., Mizin G.G., Shultezkaya L.V., Simakov B.V., Furin G.G. Synthesis of perfluoroethylcyclohexene, its alkoxy and diakylamino derivates and their electrochemical fluorination. / Abstracts of 10-European Symposium on Fluorine Chemistry, Sept. 20-25, 1992, Padua, Italy. Abstracts.B7;J.FluorineChem., 1992.v.58. р.185.
209. Молдавский Д. Д. Перфторорганические соединения: Получение прямым фторированиемуглеводородов. Дисс. докт. хим. наук, 2002, РНЦ Прикладная химия, 268с.
210. Суворов Б.В., Букейханов Н.Р. Окислительные реакции в органическом синтезе. 1978г. М., Химия, 197с.
211. Семенова Г.А., Лейтес И.Л., Аксельрод Ю.В., Маркина М.И., Сергеев С.П.,Харьковская Е.Н. Каталитические и адсорбционные методы очистки газов от сернистых соединений. –Очистка технологических газов. 1977г, М., Химия, 1977, с. 35-37.
212. Панов Г.И. Закономерности гетерогенно-каталитической активации двухатомных молекул (N2, О2, Н2). Новые каталитические системы активации молекулярного азота. Дисс. докт. хим. наук, 1986г. Новосибирск, 374с.
213. Авторское свидетельство ЧССР 220740. Способ получения тетрахлортетрафторпропана. –Паллета О., П. Свобода, Л. Стефан, Я. Квисала. –Опубл. 15.12.85. РЖ Химия, 1986, ІІН24.
214. Патент Японии 60-116637. Получение фторметана. –Такаяма С., Мэйраку Ф., Такоити А., Ковасаки Х. –Опубл. 24.06.85. РЖ Химия, 1986, 9Н16.
215. Марголис Л.Я. Окисление углеводородов на гетерогенных катализаторах. 1977г. М., Химия, с. 296.
216. Каваев М.М., Запорин А.П., Клещев Н.Ф. Каталитическое окисление аммиака. 1983г. М., Химия, с. 31.
217. Технология катализаторов. 1974г. Л.,Химия,с. 138-140.
218. Patent US 2760997. Process for the production of chlorotrifluoroethylene by passing a mixture of trichlorotrifluoroethane and hydrogen through an unobstructed iron tube. / Rucker J. T., Stormon D. B. –Publ. 28.08.56.
219. Patent US 2615925. Preparation of olefinic compounds. / Bordner C.A. –Publ. 28.10.52.
220. Patent US 2704777. Preparation of halogenated olefines / Clark J.W. –Publ. 22.03.55.
221. Patent GB 698386. Improvement in preparation of halogenated olefines / Clark J. W. –Publ. 14.10.53.
222. Patent DE 1044800. Verfahren zur Herstellung von Chlortrifluorathylen / Clark J. W. –Publ. 27.11.58.
223. Patent US 2685606. Preparation of halogenated olefines / Clark J. W. –Publ. 03.08.54 C.A. 48, 127881 (1954).
224. Patent US 2697124. Dehalogenation of fluorohalocarbons / Russel M.M. –Publ. 14.12.54 C. A. 50, 2650 (1956).
225. Patent US 2864873. Production of 1,1,2-trifluoro-2-chloroethylene / Miller C. B., Smith L. B. –Publ. 16.12.58.
226. Патент США 3043889. Получение фтористого этилена. / Смит Л.Б., Вулф К. –Опубл. 10.07.62. РЖ Химия, 1964, 3Н24.
227. Патент Германии 1186847. Получение 1-хлор-1,2,2-трифторэтилена. / Смит Л.Б., Вульф К. –Опубл. 4.10.65, РЖ Химия, 1966, 13Н26.
228. Патент США 3333011. Получение хлортрифторэтилена./Анелло Л.Г., Вульф К. –Опубл. 25.07.67 РЖ Химия, 1969, 14Н30.
229. Патент Японии 60-185734. Получение трифторхлорэтилена. / Моримото Такэси, Морикава Санэсукэ, Фунаяма Кэйди. –Опубл.21.09.85, 19Н37.
230. Европейский патент 0053657. Способ получения хлортрифторэтилена и трифторэтилена. / Канинхэм У.Д., Плэкорз Р.Е., Смит Э.М. –Опубл. 16.06.82. Изобр. в СССР и з/р., вып. 57, № 15, с. 83.
231. Патент Японии 43-8454. Способ получения трифторэтилена. / Саката Хиросукэ. –Опубл. 02.04.68. РЖХимия, 1969, 3Н36.
232. Патент США 2913400. Катализатор из окиси молибдена для конверсии углеводородов. / Буртон У.П., Лефрэнкойс Ф.А., Риблет Е.У. –Опубл. 17.11.59, РЖХимия, 1961, 9К156.
233. Патент США 3354233. Производство тетрафторэтилена. / Анелло Л.Г., Вульф К. –Опубл. 21.11.67, РЖ Химия, 1969, 9Н32.
234. Patent DE 1270547. Verfahren zur Herstellung von Tetrafluorathylen/ Anello Z. G., Wolf C. –Publ. 30.01.69.
235. Патент США 3505417. Дегалоидирование фторгалогенуглеродов. / Гарднер Л.Е. –Опубл. 07.04.70, РЖ Химия, 1971, 11133.
236. Патент США 3636173. Способ каталитического дегалоидирования./ Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н21.
237. Патент США 3789016. Катализатор гидродегалоидирования. / Гарднер Л.Е. –Опубл. 29.01.74. РЖ Химия, 1975, 2Н217.
238. Патент США 3636172. Дегалоидирование фторгалоидуглеводородов. / Гарднер Л.Е. –Опубл. 18.01.72, РЖ Химия, 1972, 21Н20.
239. Патент США 2900423. Производство перфторпропилена. / Смит Л.Б. –Опубл. 18.08.59. РЖ Химия, 1961, 7Л44.
240. Патент США 2734090. Получение фтористого винилидена. / Кафи Д.Д., Миллер Ч.Б. –Опубл. 07.02.56, РЖ Химия, 1957, 12, 42313.
241. Патент США 3404180. Получение арбонилфторида. / К.К. Лестер. –Опубл. 1.10.68. –РЖ Химия, 1969, 23Н92.
242. Патент Франции 1460246. Способ получения окиси тетрафторэтилена. / Эдисон. –Опубл. 17.10.66. –РЖ Химия, 1968 ІІНІ24.
243. Соколов Л.Ф. Исследование и разработка технологии процессов окисления фторолефинов. –
Дисс. докт. хим. наук. –Ленинград, ВНИИСК, 1979.
244. Авторское свидетельство СССР 190555. Способ получения фтористого водорода. / Гольдинов А.Л., Романов Е.И., Захаров В.Ю. и др.
245. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Термическое обезвреживание фторорганических отходов в водородо-воздушном пламени на опытно-
промышленной установке. –Отчет предприятия п/я А-
1619, 1982, инв. № 887, 18с.
246. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Катализ и инициирование деструктивного окисления, тетрафторэтилена молекулярным
кислородом. Получение карбонилфторида. –Отчет предприятия п/я А-1619, 1983, инв. № 1110, 21с.
247. Патент ГДР 213342. Способ получения фторсодержащих галоидуглеводородов со средней степенью фторирования. / Л. Ингэбург, К. Рэйнфред, М. Дитэр. –Опубл. 24.12.85. –РЖ Химия, 1986, 21Н17.
248. Патент ГДР 203314. Катализатор получения хлорфторметана в газовой фазе. / И. Лелид, Р. Каден, Д. Мросс. –Опубл. 19.10.83. –РЖ Химия, 1984, 17Н181.
249. Патент США 4504686. Способ получения 1,1,2-трифтор-2-хлорэтилдифторметилового эфира. / С. Таками. –Опубл. 12.03.85. –РЖ Химия, 1986, 8Н9.
250. Патент Японии 55-85564. Получение β-трифторметилхлорпиридинов./ Р.Нишима, К. Рудзикама, И.Йокомити, Я. Цудзии, С. Нисимура. –Опубл. 27.06.80. –РЖ Химия, 1981, 19Н177.
251. Патент США 4139568. Способ получения метилфторида. / В. Даниэль, М. Либбс. –Опубл. 13.02.79. –РЖ Химия, 1979, 18Н18.
252. Патент США 4145368. Получение 1,1,1-трифтор-2,2-дихлорэтана. Р. Свинэй, С. Бернард. –Опубл. 20.03.79. –РЖ Химия, 1979, 20Н20.
253. Патент Франции 1453510. Способ очистки дихлортетрафторэтана. / Корп. Электрохимия. –Опубл. 16.08.66. –РЖ Химия, 1968, 11Н39.
254. Виленчик Я.М., Якурнова Г.И., Остапенко Э.А. Синтез трифторацетилфторида на основе окиси тетрафторэтилена. –Тр. ГИПХ, 1985, № 101, с. 147-149.
255. Патент Японии 58-35978. Способ получения пентафторпропионилфторида. / А. Йонотаси, А. Йосио. –Опубл. 08.05.83.-Изобретения в СССР и за рубежом, 1984, вып. 57, № 8, ч.2, с. 130.
256. Патент Японии 58-38231. Получение пентафторпропионилфторида./ И. Йоносукэ, А. Йосио, С. Хироюки. –Опубл. 05.03.83.-РЖ Химия, 1984, 5Н46.
257. Патент США 3250808. Фторсодержащие простые эфиры. / Е.Ф. Мур, А. Мильян, Г. Элеутерио. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н82.
258. Патент США 3250807. Фторсодержащие карбоновые кислоты с эфирными группировками. / Ч.Г. Фритц, Е.Ф. Мур. –Опубл. 10.05.66. –РЖ Химия, 1967, 17Н84.
259. Патент США 3242218. Способ получения фторуглеродных полиэфиров. / В.Миллер. –Опубл. 22.03.66. –РЖ Химия, 1967, 12С232.
260. Патент ГДР 78376. Способ полимеризации и сополимеризации окиси гексафторпропилена в присутствии гексафторпропилена. / Г. Лотхар. –Опубл. 12.12.70. –РЖ Химия, 1971, 21 С. 361.
261. Патент США 3125599. Полимеры окисей фторуглеродных соединений. / Д. Ворнелл. –Опубл.17.03.64. –РЖХимия, 1965, 15С. 277.
262. Patent US 3291843. Fluorinated vinilethers. / Fritz C.G., Selman S., -Publ. 13.12.66.C.A. 1967, 66, 37427 h.
263. Патент США 4377717. Способ получения перфтор-2-метилпентена-2. / Л.Анелло, Р. Свинэй. –Опубл. 22.03.83. –РЖ Химия, 1983, 24Н1311.
264. Кнунянц И.Л., Фокин А.В., Чебурков Ю.А. Четвертый международный симпозиум по химии фтора в Истес-
Парк (Колорадо, США) –ЖВХО им. Д.И.Менделеева, т. 13, № 3, с. 311-321.
265. Патент США 3449389. Первичные фторзамещенные алкоголяты. / Д. Ворнелл. –Опубл. 10.06.69. –РЖ Химия, 1970, 18Н39.
266. Патент США 3322826. Полимеризация гексафторпропиленэпоксида./ Е.Ф.Мур –Опубл. 0.06.67. –РЖ Химия, 1968, 18С 296.
267. Патент Японии 57-45132. Получение перфтор-2-метил-3-оксагексаноилфторида. / Я. Масаки, М. Сэйдзи. –Опубл. 13.03.82. –РЖ Химия, 1983, 5Н70.
268. Патент США 3274293. Фторсодержащие простые эфиры. / С.Стенлей. –Опубл. 20.09.66. –РЖ Химия, 1968, 5Н161.
269. Патент Германии 2756919. Способ демеризации окиси гексафторпропилена. / Г. Кюхнэ, Ф. Геллер. –Опубл. 05.07.79. –РЖ Химия, 1980, 14Н22.
270. Патент Германии 2924385. Способ димеризации окиси гексафторпропилена. / Г. Кюхнэ. –Опубл. 07.08.80. –РЖ Химия, 1981, 23Н36.
271. Кинле Х., Бадер Э. Активные угли и их промышленное применение. –Л., Химия, 1984, с. 185-192.
272. Патент США 3321515. Способ получения фторированных карбонильных соединений. / Ф.Мур, С.Мильян. –Опубл. 23.05.67.-РЖ Химия, 1968, 15Н48.
273. Беккер Р.А., Асратян Г.В., Дяткин Б.Л. О роли пространственных факторов в реакциях полифторированных α-окисей с аминами. –ЖОрХ, 1973, т. 9, вып. 8, с. 1644-1648.
274. Кнунянц И.Л., Шокина В.В., Тюленева, В.В., Чебурков Ю.А., Аронов Ю.Е. Производные перфторизомасляной кислоты. –Изв. АНСССР,сер.хим., 1966, № 10,с. 1831-1833.
275. Sianesi D., Passety A., Tarli F., The Chemistry of hexafluoropropene epoxide. –J. Org. Chem., 1966,v. 31, № 7,p. 2312-2316.
276. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Инициирование процесса сополимеризации тетрафторэтилена и гексафторпропилена перфторированными диацильными перекисями, синтезированными на основе окиси гексафторпропилена. –Отчет предприятия п/я А-
1619, 1985, инв. № 1154, 28с.
277. Ашмор. Катализ и инициирование химических реакций. М.: Мир, 1966, с. 62.
278. Патент США 3213134. Дециклизация фторированных циклических эфиров. / Д. Морин. –Опубл. 19.10.65. –РЖ Химия, 1967, 9Н60.
279. Патент США 4302608. Способ изомеризации окиси гексафторпропилена до гексафторацетона. / Э.Скури, Д. Милз. –Опубл. 24.11.81. –РЖ Химия, 1982, 20Н56.
280. Патент Японии 58-62131. Получение гексафторацетона. / О. Йосио, А. Йосимаса, М. Макисукэ. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н30.
281. Патент Японии 58-62130. Получение гексафторацетона. / А. Йосимаса, М. Макисукэ, С. Юкио. –Опубл. 13.04.83. –РЖ Химия, 1984, 12Н29.
282. Авторское свидетельство СССР 740741. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова, Г.И. Лекомцева, Л.П. Заякина, А.П. Харченко. –Опубл. 15.06.80. –РЖ Химия, 1980, 24Н67.
283. Авторское свидетельство СССР 859348. Способ получения гексафторацетона. / Я.М. Виленчик, Г.И. Якурнова. –Опубл. в Б.И., 1981, № 32, РЖ Химия, 1982, 21Н12.
284. Патент США 4238416. Способ изомеризации фторированных соединений. / Д. Кодуо. –Опубл. 09.12.80. –РЖ Химия, 1981, 17Н65.
285. Киперман С.Л. Введение в кинетику гетерогенных каталитических реакций. М.: Наука, 1964, с. 401.
286. Тюльга Г.М., Губанов В.А., Попова И.С., Петрухно Л.А., Болховец Б.М., Тройчанская П.Е., Долгопольский И.М. Изучение реакции фторангидридов перфторкарбоновых кислот с фторидами щелочных металлов. –ЖОрХ, 1973, т. 14, вып. 11, с. 2339-2346.
287. Коншин А.И., Губанов В.А. Об оценке эффективности способа очистки хладоновых растворов перфторполиоксаметиленацетилфторидов. –Отчет предприятия п/я В-8415.
288. Авторское свидетельство СССР 188404. Фторсополимер винилиденфторида с 3,6,8,10,12-пентаоксаперфтортридеценом для термоморозоагрессивостойких изделий. / Грейс А.М., Ершов А.Е., Захаров В.Ю. и др.
289. Карцов С.В., Валов П.И., Соколов Л.Ф., Соколов С.В. Роль поверхности реакционного сосуда в процессе жидкофазного окисления гексафторпропилена. –Изв. АН СССР, сер. хим., 1978, № 10. с. 2268-2272.
290. Авторское свидетельство 229185. Способ получения перфторполиоксаметиленацетилфторидов. / Захаров В.Ю., Гольдинов А.Л., Боровнев Л.М. и др.
291. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика изменения состава газовой и жидкой фаз в промышленном реакторе синтеза окиси гексафторпропилена в ходе операции окисления. –Отчет предприятия п/я А-1619, 1983, инв. № 1089, 10с.
292. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Динамика образования перфторполиоксаметилен ацетилфторидов в процессе жидкофазного окисления гексафторпропилена кислородом на промышленной установке. –Отчет предприятия п/я А-1619, 1985, инв. № 1165, 22с.
293. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Окисление гексафторпропилена молекулярным кислородом в присутствии 1,2-дибромтетрафторэтана (хладона-114В2). Получение окиси гексафторпропилена. –Отчет предприятия п/я А-1619, 1982, инв. № 1072, 57с.
294. Авторское свидетельство СССР 1128436. Пламягасящая смесь. / Боровнев Л.М., Гусенков М.В., Жукова В.А. и др.
295. Кравченко Н.Н., Попов В.А., Ершов Ю.А., Померанцева Э.Г. Некоторые особенности фотолиза перфторированных полиэфиров. –ВМС, т. 23, № 3, 1981, с. 180-183.
296. Заявка СССР 3011822. Способ регенерации хладона-113. / Паниткова Е.С., Беренблит В.В., Сенюшов А.Н. и др.
297. Зейфман Ю.В., Тер-Габриэлян Е.Г., Ганбарян Н.П., Кнунянц И.Л. Химия перфторизобутилена. –Успехи химии, 1984, т. 53, вып. 3, с. 431-461.
298. Кнунянц И.Л., Шокина В.В., Кулешова А.Д. Присоединение галоидводородов к фторолефинам. –Изв. АН СССР, сер. хим., 1960, № 9, с.1693-1695.
299. Постовой С.А., Мысов Е.И., Зейфман Ю.В., Кнунянц И.Л. Нуклеофильная конденсация октафторизобутилена с тетрафторэтиленом. –Изв. АН СССР, сер. хим., 1982, № 7, с. 1586-1590.
300. Патент США 3389187. Димер перфторизобутилена. / Д.Л. Миллер. –Опубл. 18.06.68. –РЖ Химия, 1969, 17Н3211.
301. Кнунянц И.Л., Герман Л.С., Дяткин Б.П. Реакции фторолефинов. Сообщение ІІ. Взаимодействие соединений ряда перфторизобутилена с аминами и аммиаком. –Изв. АН СССР, отд.хим.н., 1960, № 2, с. 221-230.
302. Патент Японии 53-73504. Удаление октафторизобутилена. / Курода Такэси, Фурукава Йосики, Мацуоха Ну. –Опубл. 30.06.78. РЖХимия, 1979, 10Н2011.
303. Дорогимский В.А., Коломиец А.Ф., Сокольский Г.А. О реакции октафторизобутилена с этиленгликолем и глицерином. –ЖОрХ, 1983, т.19, вып. 8, с.1762-1763.
304. Авторское свидетельство СССР 129653. Способ получения гексафторизомасляной кислоты. / Кнунянц И.Л., Чебурков Ю.А., Красуская М.П. –РЖ Химия, 1961, 9Л75.
305. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Промышленные испытания и внедрение способа очистки газов пиролиза тетрафторэтилена от перфторизобутилена. –Отчет предприятия п/я А-1619, 1981, инв. № 1048, 34с.
306. Гольдинов А.Л., Боровнев Л.М., Захаров В.Ю. и др. Увеличение степени использования сырья в производстве гексафторпропилена путем более полного извлечения октафторциклобутана из кубовой фракции. –Отчет предприятия п/я А-1619, 1982, инв. № 1063, 16с.
307. Авторское свидетельство СССР 180854. Способ получения гексафторпропилена. / Боровнев Л.М., Виноградова А.П., Голубев А.Н. и др.
308. Боровнев Л.М., Голубев А.Н., Захаров В.Ю. и др. Комплексная переработка кубовой фракции производства тетрафторэтилена. –Отчет предприятия п/я А-1619, 1985, инв. № 15/107 ДСП, 17с.
309. Авторское свидетельство СССР 1336492. Способ получения 2-хлортетрафторэтилфторсульфата. / Фокин А.В., Рапкин А.И., Татаринов А.С. и др.
310. Авторское свидетельство СССР 1184808. Способ получения монофторсульфата брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
311. Авторское свидетельство СССР 1239092. Способ получения трис(фторсульфата) брома. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
312. Заявка СССР 3982041. 3-(Полифторпропионамидо) пропил-2-оксиэтилдиметиламмоний хлориды, обладающие гипотензивным действием. / Фокин А.В., Ковалев Г.В., Мазанов Л.С. и др.
313. Заявка СССР 3982040. N-Полифторацильные производные γ-амномасляной кислоты, обладающие гипертензивным действием. / Фокин А.В., Ковалев Г.В., Мухина Н.В. и др. Пол. реш. от 26.09.86.
314. Автотское свидетельство СССР 1334655. 3-(ω-Хлорперфторалканоиламидо)пропил-диметил-2-оксиэтиламммоний хлориды в качестве поверхностно-активных веществ. /Фокин А.В., Рапкин А.И., Студнев Ю.Н. и др.
315. Авторское свидетельство СССР 196359. Способ получения ω-хлорперфторкарбоновых кислот и эмульгатор. / Фокин А.В., Студнев Ю.Н., Рапкин А.И. и др.
316. Патент Германии 3034549. Способ получения перфторированных фторангидридов карбоновых кислот. / Вернер Ш. –Опубл. 29.04.82, –РЖ Химия, 1983, 17Н5711.
317. Промышленные катализаторы и носители. Справочник. –Новосибирск, 1972, с. 246.
318. Т. Ван дер Плас. Текстура и химия поверхности углеродных тел. В кн.: Строение и свойства адсор
бентов и катализаторов. –М.: Мир, 1973, с. 436-441.
319. Патрик С. Успехи химии фтора. –М., Л., Химия, 1964, т.1, № 2, с. 336-379
Материал рекомендован к публикации членом редколлегии С.М. Игумновым
Fluorine Notes, 2014, 97, 1-2
